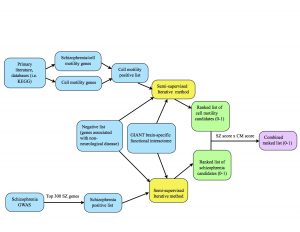
This previous week we put our iterative algorithm to the test. Alex and I each ran our positive (and negative) lists for 150 iterations using the 0.200 threshold GIANT brain network. As a reminder, the 0.200 threshold network is a trimmed version of the GIANT network where all the edges have probability scores of 0.200 or greater.
I used all 541 of my positives, a combination of two sublists: cell motility genes and cell motility genes implicated in schizophrenia. In the upcoming weeks, I will experiment with running these two lists separately and seeing how the results are affected.
I also used Alex’s list of negatives, which are genes expressed in other organs but not the brain. We will need to closely consider whether it is wise to use this set of negatives alongside cell motility positives.
Even with these two potential issues, our preliminary lists of candidates looked promising – for example, one gene we found is Hes Related Family BHLH Transcription Factor With YRPW Motif 2, also know as HEY2. HEY2 was not only a schizophrenia positive but was also moderately high in the list of cell motility candidates, with a score of 0.56. Another gene we uncovered was 1-Acylglycerol-3-Phosphate O-Acyltransferase 4, or AGPAT4. This was not in either list of positives, but it was given the maximum score of 1.0 in each set of candidates. Both of these genes point to the different ways we can analyze our results and what it means to be a promising candidate – do we care about genes that are positives in the schizophrenia set and are determined by the algorithm to likely be involved in cell motility or do we care about genes that are determined by the algorithm to be likely involved in cell motility and schizophrenia? Do we care about both? What is the difference? Is one more promising than the other? In the upcoming weeks, we will be figuring out these questions in order to refine our list of candidates.
We will also be extending the time that our algorithm runs. 150 iterations may seem like a lot, but cutting the algorithm short may lead to “stunted” results. The iterative method can be thought of as dyes from the positive and negative nodes spreading throughout the network, staining the surrounding nodes in a way that reflects their distances from the positives and negatives. If the iterations are stopped before they converge (i.e. subsequent iterations don’t change the scores, or color of the node), then the color of the nodes might be closer to the color of the positives and negatives than they should be. Our next step will be to let it run for 1500 iterations and see how our results our affected.
Out of my own curiosity, I also decided to see if all of the cell motility positives were in the GIANT network, which turned out to be false. There are about 40 genes (out of 541) that are not in the GIANT network. However, I don’t believe this to be a problem; if there are positive cell motility genes not in the GIANT network, it is probably because not all genes involved in cell motility are expressed in every type of cell and are therefore not relevant in a brain-specific network.