Molecular cloning with Gibson Assembly
Molecular cloning is a technique established in the early 1970s that made introducing specific genes into a host possible [1]. This significantly advanced research because it allowed us to isolate and study individual genes. One method cloning has made possible is protein purification. By introducing a gene to a bacterial vector, the encoding protein is able to be overexpressed and then subjected to biochemical analyses such as crystallization or enzymatic assays to elucidate the function of the protein [2].
I am using this exact technique in my senior thesis at Reed College to study CG11811, a protein novelly indicated as a non-muscle myosin II (NMII) regulator. NMII is an actin binding protein that when activated, constricts actin filaments to change cell shape [3]. Our previous studies found that when CG11811 was depleted using RNAi, there were decreased contractility events. However, we do not know how exactly CG11811 regulates NMII. Therefore, I am cloning CG11811 into a strain of E. coli designed to mass produce proteins in order to purify it and run enzymatic assays to reveal some of its biochemical properties. The first step of this cloning process is creating the recombinant DNA plasmid to transform into the bacteria.
A very efficient technique for this purpose is Gibson Assembly, designed by Dr. Daniel Gibson in 2009 [4]. With Gibson cloning, any two sequences (the gene of interest and the vector) can be joined together without the need for restriction enzyme sites (Figure 1A). The basis of this technique is to design two sets of primers that overlap the gene and the vector (Figure 1B). When these sets of primers are used in PCR, the vector has a section at the end that is complementary to the insert and the insert has a section at the end that is complementary to the vector (Figure 1B). These two PCR products are then combined in the Gibson reaction with an exonuclease that will chew back the overlaps to create “sticky” ends that will anneal to each other, polymerase to fill in any gaps in base pairs in the overlaps, and ligase to join the fragments together (Figure 2). The resulting product is a contiguous plasmid that can then be transformed into the protein expression E. coli to get mass quantities of CG11811.
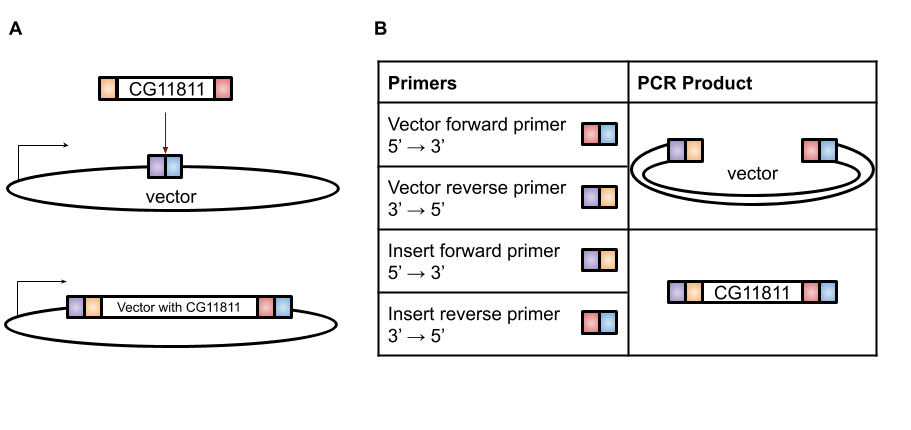

Citations
1. Jackson, D. A., Symons, R. H., & Berg, P. (1972). Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 69(10), 2904–2909. doi:10.1073/pnas.69.10.2904
2. Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in microbiology, 5, 172. doi:10.3389/fmicb.2014.00172
3. Martin, A. C., Kaschube, M., & Wieschaus, E. F. (2009). Pulsed contractions of an actin–myosin network drive apical constriction. Nature, 457(7228), 495–499. https://doi.org/10.1038/nature07522
4. Gibson, D. G., Young, L., Chuang, R.-Y., Venter, J. C., Hutchison, C. A., & Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6(5), 343–345. https://doi.org/10.1038/nmeth.1318