by Nanati Safawo
Schizophrenia is a neurodevelopmental disorder and it is caused when the brain is not formed normally in early life. Some of the symptoms can include delusions, hallucinations, disorganized speech, trouble with thinking, and lack of motivation. Emerging studies are showing that Schizophrenia can impede neuronal network formation and neurodevelopmental processes such as differentiation and migration(Berreta, 2016). Since The Extracellular Matrix (ECM) plays a key role in the regulation of cell differentiation and migration and axonal outgrowth and guidance (Bandtlow and Zimmermann, 2000), we predict that aberrant interaction between the neurons and(ECM) could cause disruption observed in Schizophrenia.
Our project is studying the neuronal-ECM relationships, and how ECM signaling dictates the differentiation of neuroblasts, axonal extensions, and neuronal architecture of neurons and is comparing these developments in terms of their axonal extension across different substrates of ECM and Concanavalin A (Con A). Our long-term goal is to understand what could cause the aberrant interaction between neurons and ECM.
ECM is a large network of proteins and other molecules that surround, support, and give structure to cells and tissues in the body. It plays a critical role in dictating cellular behavior. In addition, ECM also helps cells attach to, and communicate with nearby cells and plays an important role in cell growth, movement, and other cell functions. Con- A, our other substrate, is a lectin that does not engage ECM receptors and thus does not convey vital signaling information to neurons. Con A is used because the cells stick well but there is no real biologically significant signaling that is happening. Hence using it as a substrate could act as a control to aid our understanding of how ECM dictates neuronal behavior and how neurons across species interact with their extracellular environment.
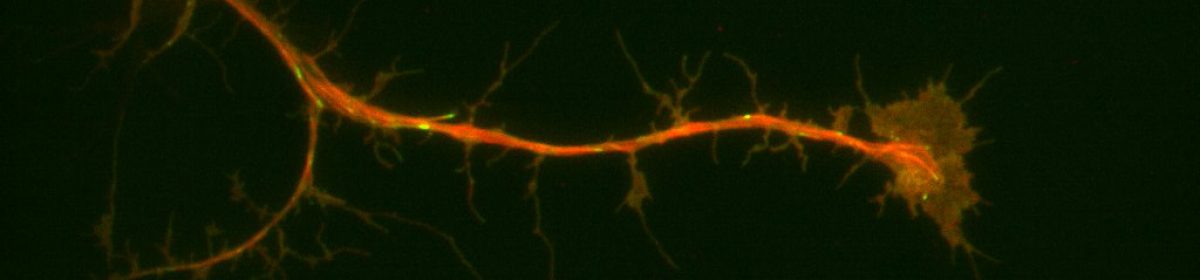
Neuronal progenitor cells from the brain of Drosophila melanogaster (fruit flies) is our preferred system to study the neuronal-ECM relationships. During neurogenesis, neuroblasts undergo a series of divisions either producing more of themselves or differentiating into neurons or glia1. Brains from larvae, which are enriched in neuroblasts, were removed and cultured for days. This system is ideal for detailed, high-resolution microscopy and exploring signaling from the ECM since it has formerly led to fundamental discoveries furthering our understanding of neurons and neurogenesis. Additionally, purified ECM is more biologically relevant because when coupled with compliant substrates, it mimics the softer environments of brains representing a powerful system allowing us to probe ECM-neuronal interactions from several different axes. Our preliminary data suggested that neurons plated on nECM behave differently than those plated on Con A. We collected Neuroblasts from third-instar larval with the genetic background of (Prospero-GAL4xUAS-EB1::EGFP; UAS-Jupiter::mCherry) and plated them on the substrate of either ECM or ConA. We used TIRF microscopy for imaging and observing the development of our neurons. In parallel with our predictions, we replicated former lab findings demonstrating that microtubule dynamics and axonal extensions increased in neurons plated on ECM compared to con A.
Another line of inquiry we pursued was using the UAS-RNAi fly line in combination with the inscrutable-Gal4 driver to deplete the major adhesion molecule, Syndecan, and observe neuroblast behavior when plated on ECM and Con A. Interestingly, the results we got from these were quite surprising. The neurons plated on ECM did not show much growth and had axonal extensions. The neurons were unable to sense the signaling from the ECM and did not develop or show axonal extensions; instead, they either died or showed minimal development. This result suggests that Syndecan might contribute significantly to the regulation of the neuronal-ECM interaction. Furthermore, it could have an impact on the axonal growth of neurons on ECM, neuronal differentiation, and overall neuronal development which in turn might contribute to the cause of SZ. We are currently working on replicating these results and I am excited to see what we will find out.
Berretta, S. (2012a, March). Extracellular matrix abnormalities in schizophrenia. Neuropharmacology. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3234338/#R18
Berretta, S. (2012b, March). Extracellular matrix abnormalities in schizophrenia. Neuropharmacology. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3234338/#R18
Stephen J Glatt, Stephen V Faraone, Ming T Tsuang. (n.d.). Is Schizophrenia A Neurodevelopmental Disorder?. Academic.oup.com. https://academic.oup.com/book/40600/chapter-abstract/348206860?redirectedFrom=fulltex
‘Polarizing’ Adventures: Microtubules Lead the Way
-By Nicole Chan
Cell migration depends on the dynamics of the actin and microtubule cytoskeleton. Classically, the actin cytoskeleton is the “engine” of migration, driving cell protrusion. The microtubule cytoskeleton has long been hypothesized to play the role of “compass”, dictating the direction of migration. Another key element to cell migration is focal adhesions, plague-like structures made up of some 120 different proteins that connect the cytoskeleton to the extracellular matrix (ECM). To put it all together briefly, protrusion from the actin cytoskeleton lays the groundwork for the formation of focal adhesions which mature and strengthen the more the cell physically pulls on them. Meanwhile, microtubules direct where protrusions occur and effectively steer the cell. Focal adhesion expression and dynamics also correlate strongly with metastasis in human epithelial cancers which is why studying them has important ramifications for human health. A recent publication on focal adhesions from the Applewhite lab has found that disruption of microtubule polarity (via depletion of the major actin-microtubule cross-linking factor known as Short stop of Shot) results in faster focal adhesion turnover. This ultimately results in faster cell migration (Zhao, AJ., Montes-Liang J., et al, 2022). If disruption of microtubule alignment with actin and focal adhesions led to faster focal adhesion turnover, we wondered what would happen to focal adhesion dynamics if microtubule polarity was forced in the direction of migration. Drosophila cells are a somewhat naive system to test these ideas as they lack an active centrosome during interphase. By expressing a constitutively active Polo Kinase construct, we can activate the centrosome, leading to the formation of a microtubule organization center which then can force the microtubules, putatively in the direction of migration. Results from Summer 2023 research, indicate a presence of a centriole-like structure that organizes the microtubules. It is our hypothesis that this forced polarity will result in cell-matrix adhesions that will last longer, but it is not known how this impacts cell migration. The primary question driving this project is how Drosophila cell-matrix adhesion dynamics behave in cells with or without active centrosomes. However, due to the short timeline, I will be aiming to quantify cell-matrix adhesion dynamics in cells with and without centrosomes.
Zhao, A. J., Montes-Laing, J., Perry, W. M. G., Shiratori, M., Merfeld, E., Rogers, S. L., & Applewhite, D. A. (2022). The Drosophila spectraplakin Short stop regulates focal adhesion dynamics by cross-linking microtubules and actin. Molecular biology of the cell, 33(5), ar19. https://doi.org/10.1091/mbc.E21-09-0434
Thinking Outside the Cell: A Neuroscientist’s Guide to the Extracellular Matrix and Focal Adhesions
Introduction
Biological organisms are incredibly complex systems, capable of completing a wide array of actions and behaviors, mostly through the creation of specialized tissues which work in synchrony to produce these complex actions. A simple example would be locomotion, which requires multiple tissue types such as bone, muscle, and connective tissue. Each tissue’s properties are integral to its function, and without this variety in tissue type and properties, many complex biological behaviors could not exist. These properties arise not only from the cells within the tissues, but from the matrix of proteins on which they grow, known as the extracellular matrix (ECM). This matrix is a collection of proteins and small molecules, whose properties can vary greatly based on its exact composition. (Manou et al., 2019). Cells communicate back and forth with the ECM through protein complexes known as focal adhesions (FAs) (Wehrle-Haller et al., 2012). In this review I will provide a basic introduction to the ECM and FAs to highlight how cell-ECM interactions are a critical component of proper tissue differentiation and function, with a particular focus on how these interactions are relevant in a neuroscience setting. Rather than provide a fully comprehensive breakdown on the topic, this review should serve to provide a framework for understanding how these factors may affect cells, and expand the neuron-centric neuroscience curricula present at many institutions.
Basics of the Extracellular Matrix
In vivo, cells are not just packed in an array, but rather reside on a complex matrix of proteins which provides both the necessary “stuff” for cells to live on, and acts as a transmitter of information for the cells within this extracellular matrix (ECM) (Manou et al., 2019). This matrix is composed partially of proteins synthesized by the cell, which are interconnected with each other, but also connected to the cells within the matrix. This physical network allows for bidirectional regulation and signaling between cells and the ECM which enables biochemical information to move throughout this network (Bonnans et al., 2015). This matrix also dictates much of the chemical and physical properties of the tissue it composes, which is determined mostly by the concentrations of each ECM component. Here I will discuss the major components which could be considered the foundation or skeleton of the ECM and how their concentration affects the properties of the matrix as a whole.
The most abundant protein in most vertebrate ECMs is the collagen superfamily, containing 28 different types, which serve to create a rigid filamentous network (Sorushanova et al., 2019). Collagens are generally identified by their triple helical structure, which imparts a resistance to stretching to the collagen strand, making it a skeleton upon which the rest of the ECM can be built (Shoulders and Raines, 2009). The concentration of collagen is proportional to the rigidity of the medium, with higher concentrations being more rigid (Kadler et al., 2007). Due to its prevalence, there are many examples of collagen in vivo, but a possibly unappreciated role is in thrombosis and hemostasis, where collagen serves as an insoluble scaffold for other vascular proteins and cells (Farndale et al., 2004).
Proteoglycans are another large component of the ECM, and are composed of a core protein which holds multiple glycosaminoglycans (GAGs), with the exact quantity and composition varying depending on the specific proteoglycan, which impart a large negative charge to the molecule (Theocharis and Karamanos, 2019). This charge causes counterions and associated water molecules to be “absorbed” by the proteoglycans, imparting a more gel-like texture, which provides some resistance to compression (Zhao et al., 2013). The large variation of possible GAGs that compose a proteoglycan makes it useful in regulating tissue development. This is notable in the brain, where highly regulated spatio-temporal expression of proteoglycans plays a critical role in proper brain development, migration, axonal pathfinding, synaptogenesis, and plasticity (Schwartz and Domowicz, 2018).
A finer, mesh-like structure is also present in the form of laminins, a cross-shaped trimeric protein with binding sites for collagen and other compounds (Durbeej, 2010). Due to its relatively smaller size, laminin is the predominant anchor for cells to the ECM and as such plays a critical role in ECM morphogenesis (Miner and Yurchenco, 2004). This has been observed in the nervous system, with microglial morphology being affected by laminin in vitro (Tam et al., 2016).
These proteins interact with each other and cells through the help of fibronectins, a covalently bonded symmetrical dimer with a large array of binding sites which can link the ECM to a cell or other ECM components (Pankov and Yamada, 2002). Fibronectin can often move along fibrillar structures to help cell migration and matrix rearrangement (Dallas et al., 2006). Knocking out fibronectin in mouse embryos was lethal before embryogenesis, but homozygous fibronectin mutants showed widespread defects in mesoderm, neural tube, and vascular development. The observed deformities in neural tube and vascular structure seem to reinforce fibronectin’s key role in cell adhesion and migration (George et al., 1993)
A common feature of the ECM proteins mentioned, besides proteoglycans, is the RGD loop, named after its sequence (Arg-Gly-Asp), and functions as a critical binding site for cell adhesion (Ruoslahti, 1996).
Varying concentrations of ECM compounds will alter ECM properties, such as rigidity. These changes in rigidity can be critical in cell proliferation, morphogenesis, and differentiation (Tilghman et al., 2010). While historically the brain was thought to lack an ECM, persistent research identified not only its presence, but its significance in proper brain development and maintenance as well. Neuronal ECM contains a much lower concentration of collagen relative to other ECMs, and composes 15-20% of the brain’s volume (Lei et al., 2017). Rather than collagen, the neuronal ECM is composed primarily of proteoglycans, which are thought to help synaptic plasticity (Lau et al., 2013). However, the aforementioned role of collagen in maintaining vascular integrity is conserved in the blood-brain barrier (Thomsen et al., 2017). While neurons have been shown to be able to bind directly to laminin even in the absence of fibronectin (Liesi et al., 1984), fibronectin supports neurite outgrowth and axonal regeneration (Tonge et al., 2012). Although research on neuronal ECM has steadily increased, there is still much to be uncovered.
Basics of Cell-Matrix Interactions
Focal adhesions (FAs) are multi protein complexes that serve as a transductor of information, both mechanical and chemical, across the membrane, while also functioning as an anchor or linker for the cell to the ECM (Wehrle-Haller et al., 2012). Since there is such a wide range of proteins involved in FAs, I will only be discussing some of the more critical and well-studied proteins.
Integrins are a class of transmembrane heterodimeric proteins composed of 24 unique combinations from 18 ⍺-subunits and 8 𝛽-subunits and serve as the primary transmitters of information across the membrane (Kechagia et al., 2019). Integrins adopt an inactive conformation when unstimulated in which the cytoplasmic tails are bound. Talin binding is observed to be a final common step in integrin activation, since it is critical for triggering structural change in the protein. (Tadokoro et al., 2003). Additionally, the kindlin protein also serves to facilitate integrin activation by binding to the 𝛽-subunit cytoplasmic tail. (Rognoni et al., 2016). When activated integrins can identify a wide variety of ECM compounds, which is primarily determined by the specific combination of ⍺- and 𝛽-subunits (Hynes, 2004). A common binding site integrin is the RGD loop which is notably found in both laminin and fibronectin within the ECM (Takagi et al., 2003).
Within the central nervous system, most integrin isoforms are expressed throughout the brain with some degree of variation across different regions (Clegg et al., 2003). In addition to their roles in other cells, integrins are found to play a role in synapse regulation and plasticity within neurons (Park and Goda, 2016).
Integrin’s primary connection to the actin cytoskeleton is through the previously mentioned talin, which binds to the 𝛽-subunit cytoplasmic tail of the integrin dimer. Talin contains multiple binding sites for actin, which are adjacent to a vinculin binding site (Hemmings et al., 1996). Vinculin is a protein which in addition to binding talin, can also bind actin, strengthening the connection between the two (Rosowksi et al., 2018). This vinculin binding site is only exposed when talin undergoes deformation by being stretched open (Lee et al., 2007). This reinforces talin-actin interactions when under sufficient tension by causing a vinculin-mediated increase in talin-actin interactions following the exposure of new vinculin binding sites. This is supported by cells expressing vinculin exerting far greater forces than cells lacking vinculin (Mierke et al, 2007).
Another key player in cell-matrix interactions is focal adhesion kinase (FAK), a non-receptor tyrosine kinase, a signaling molecule activated by FAs and serves as a relay of information between FAs and the rest of the cell (Tapial Martínez et al., 2020). FAK is activated when the molecule forms a complex with the FA and is stretched open when tension is present (Seong et al., 2013). When activated FAK can form a complex with c-Src which is involved in multiple signaling pathways, most of which are related to cell motility and growth (Mitra and Schlaepfer, 2006).
While the binding patterns mentioned above seem to be the dominant interactions that lead to propper FA function, most FA proteins can bind with multiple other FA proteins. Although this would suggest that the proteins are clumped together somewhat haphazardly, FA structure is actually precisely regulated (Parsons et al., 2010). Part of this regulation is due to the spatial distribution of proteins with the FA, which produces functional subregion (Schwartz, 2011). Signals from the ECM will pass through the integrin signaling layer (ISL) and are then carried through the force transduction layer (FTL) before reaching the actin regulatory layer (ARL). This nano-organization has been shown to play an important role in regulating talin-mediated vinculin activation, and is likely to do the same for many other FA protein interactions (Case et al., 2015).
Biochemical Response to ECM Properties
Cells and the extracellular matrix engage in a bidirectional relationship in which the assembly of the ECM is regulated by signaling pathways within the cell, whose activity is dependent on cell surface receptors that recognize ECM proteins. This leads to variation in ECM biochemical composition, morphology, and rigidity, imparting chemical and physical qualities which facilitate proper tissue function (Gasiorowski et al., 2013). In this section I will explore some of these relationships between cells and the ECM.
It has long been known that the ECM plays a large role in determination of cell shape (Gospodarowicz et al., 1978). This interaction is similarly dependent on bidirectional signaling, in which the ECM provides contextual cues to the cells, thereby directing their phenotype, while cells remodel the ECM, altering the context itself (Gjorevski and Nelson, 2009). Changes in cell shape are largely due to the rigidity of the matrix, and therefore plays a large role in cell differentiation (Engler et al., 2006). Multiple ECM components have been shown to have a role in regulating branching morphogenesis in various cell types through changes in rigidity and by activating signaling cascades within the cell upon binding (Rozario and DeSimone, 2009). Other factors may affect this interaction. For example, in neurons, the response to ECM rigidity is regulated by neuron-astroglia interactions (Georges et al., 2007).
These effects are not limited to morphology and morphogenesis but extend also to migration, where the ECM plays a critical role in properly directing the cell (Yamada and Sixt, 2019). Before the cell even moves, the ECM directs cell polarity, an important factor in proper cell migration (Manninen, 2015). This is accompanied by an ECM dependent regulation of migration speed (Palecek et al., 1997).
In turn, cells direct a dynamic process of ECM degradation and remodeling which ultimately determines the properties of the ECM (Lu et al., 2011). One of the primary, and possibly simplest, mechanisms for ECM degradation is with the aid of proteinases which can selectively degrade a specified ECM protein (Cawston and Young, 2009). Alternatively, ECM proteins can often be regulated by posttranslational modifications (Frantz et al., 2010). A good example of this would be collagen, which can be cross-linked either covalently or noncovalently, causing a substantial change in ECM topography and rigidity (Levental et al., 2009). These processes of ECM degradation and regulation are themselves regulated either by gene regulation or environmentally (Page-McCaw et al., 2007).
Final Notes
These complex interactions between cells and the matrix highlights how cells do not exist as isolated systems within an organism. They are constantly exchanging information with their environment, which can be transmitted to other cells in the matrix, helping to add a foundation and network for communication and coordination between cells. It is therefore important to keep in mind the ECM when conducting research in any tissue, as to account for the behavior of cells in their native environment. This is equally true in neuroscience, where neurons often receive more attention than their neighboring glial cells. Expanding our understanding of the nervous system as more than a clump of neurons, but rather as a complex system of various cell types interconnected and supported by an ECM whose own composition may be as critical in proper cognitive function and development as the more popular neuronal circuits.
References
1. Zhao, Y. et al. Proteoglycans and Glycosaminoglycans Improve Toughness of Biocompatible Double Network Hydrogels. Advanced Materials 26, 436–442 (2014).
2. Yamada, K. M. & Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 20, 738–752 (2019).
3. Wehrle-Haller, B. Structure and function of focal adhesions. Current Opinion in Cell Biology 24, 116–124 (2012).
4. Tonge, D. A. et al. Fibronectin supports neurite outgrowth and axonal regeneration of adult brain neurons in vitro. Brain Res. 1453, 8–16 (2012).
5. Tilghman, R. W. et al. Matrix Rigidity Regulates Cancer Cell Growth and Cellular Phenotype. PLoS One 5, (2010).
6. Thomsen, M. S., Routhe, L. J. & Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 37, 3300–3317 (2017).
7. Theocharis, A. D. & Karamanos, N. K. Proteoglycans remodeling in cancer: Underlying molecular mechanisms. Matrix Biol. 75–76, 220–259 (2019).
8. Tapial Martínez, P., López Navajas, P. & Lietha, D. FAK Structure and Regulation by Membrane Interactions and Force in Focal Adhesions. Biomolecules 10, (2020).
9. Tam, W. Y., Au, N. P. B. & Ma, C. H. E. The association between laminin and microglial morphology in vitro. Sci Rep 6, 28580 (2016).
10. Takagi, J., Strokovich, K., Springer, T. & Walz, T. Structure of integrin α5β1 in complex with fibronectin. https://www-ncbi-nlm-nih-gov.proxy.library.reed.edu/pmc/articles/PMC212714/ (2003).
11. Tadokoro, S. et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science 302, 103–106 (2003).
12. Sorushanova, A. et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv. Mater. Weinheim 31, e1801651 (2019).
13. Shoulders, M. D. & Raines, R. T. COLLAGEN STRUCTURE AND STABILITY. Annu Rev Biochem 78, 929–958 (2009).
14. Seong, J. et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 19372–19377 (2013).
15. Schwartz, N. B. & Domowicz, M. S. Proteoglycans in brain development and pathogenesis. FEBS Lett. 592, 3791–3805 (2018).
16. Schwartz, M. A. Super-resolution microscopy: a new dimension in focal adhesions. Curr. Biol. 21, R115-116 (2011).
17. Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715 (1996).
18. Rozario, T. & DeSimone, D. W. The Extracellular Matrix In Development and Morphogenesis: A Dynamic View. Dev Biol 341, 126–140 (2010).
19. Rosowski, K. A. et al. Vinculin and the mechanical response of adherent fibroblasts to matrix deformation. Sci Rep 8, (2018).
20. Rognoni, E., Ruppert, R. & Fässler, R. The kindlin family: functions, signaling properties and implications for human disease. J. Cell. Sci. 129, 17–27 (2016).
21. Parsons, J. T., Horwitz, A. R. & Schwartz, M. A. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 11, 633–643 (2010).
22. Park, Y. K. P. & Goda, Y. Integrins in synapse regulation | Kopernio. https://kopernio.com/viewer?doi=10.1038%2Fnrn.2016.138&token=WzEyNDAwNTEsIjEwLjEwMzgvbnJuLjIwMTYuMTM4Il0.78Am3n4-yLVXLla4lx-OkHdSqOA.
23. Pankov, R. & Yamada, K. M. Fibronectin at a glance. J. Cell. Sci. 115, 3861–3863 (2002).
24. Palecek, S. P., Loftus, J. C., Ginsberg, M. H., Lauffenburger, D. A. & Horwitz, A. F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385, 537–540 (1997).
25. Page-McCaw, A., Ewald, A. J. & Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233 (2007).
26. Mitra, S. K. & Schlaepfer, D. D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 (2006).
27. Miner, J. H. & Yurchenco, P. D. Laminin functions in tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 20, 255–284 (2004).
28. Mierke, C. T. et al. Mechano-Coupling and Regulation of Contractility by the Vinculin Tail Domain. Biophys J 94, 661–670 (2008).
29. Manou, D. et al. The Complex Interplay Between Extracellular Matrix and Cells in Tissues. in The Extracellular Matrix: Methods and Protocols (eds. Vigetti, D. & Theocharis, A. D.) 1–20 (Springer, 2019). doi:10.1007/978-1-4939-9133-4_1.
30. Manninen, A. Epithelial polarity–generating and integrating signals from the ECM with integrins. Exp. Cell Res. 334, 337–349 (2015).
31. Lu, P., Takai, K., Weaver, V. M. & Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb Perspect Biol 3, (2011).
32. Liesi, P., Dahl, D. & Vaheri, A. Neurons cultured from developing rat brain attach and spread preferentially to laminin. J. Neurosci. Res. 11, 241–251 (1984).
33. Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
34. Lei, Y., Han, H., Yuan, F., Javeed, A. & Zhao, Y. The brain interstitial system: Anatomy, modeling, in vivo measurement, and applications. Prog. Neurobiol. 157, 230–246 (2017).
35. Lee, S., Kamm, R. & Mofrad, M. Force-induced activation of Talin and its possible role in focal adhesion mechanotransduction – ProQuest. http://search.proquest.com/docview/1034928518?accountid=13475 (2007).
36. Lau, L. W., Cua, R., Keough, M. B., Haylock-Jacobs, S. & Yong, V. W. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat. Rev. Neurosci. 14, 722–729 (2013).
37. Kechagia, J. Z., Ivaska, J. & Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473 (2019).
38. Kadler, K., Baldock, C., Bella, J. & Boot-Hanford, R. Collagens at a glance | Kopernio. https://kopernio.com/viewer?doi=10.1242%2Fjcs.03453&token=WzEyNDAwNTEsIjEwLjEyNDIvamNzLjAzNDUzIl0.iBYQOEZkdX-fRDLOgfil4P3SiQ4 (2007).
39. Joanny, J.-F. & Prost, J. Active gels as a description of the actin-myosin cytoskeleton. HFSP J 3, 94–104 (2009).
40. Hynes, R. O. & Naba, A. Overview of the Matrisome—An Inventory of Extracellular Matrix Constituents and Functions. Cold Spring Harb Perspect Biol 4, (2012).
41. Hynes, R. O. The emergence of integrins: a personal and historical perspective. Matrix Biol. 23, 333–340 (2004).
42. Hemmings, L. et al. Talin contains three actin-binding sites each of which is adjacent to a vinculin-binding site. J. Cell. Sci. 109 ( Pt 11), 2715–2726 (1996).
43. Gospodarowicz, D., Greenburg, G. & Birdwell, C. R. Determination of cellular shape by the extracellular matrix and its correlation with the control of cellular growth. Cancer Res. 38, 4155–4171 (1978).
44. Gjorevski, N. & Nelson, C. M. Bidirectional extracellular matrix signaling during tissue morphogenesis. Cytokine Growth Factor Rev 20, 459–465 (2009).
45. Georges, X. J. P. C. et al. Cell Growth in Response to Mechanical Stiffness is Affected by Neuron- Astroglia Interactions. The Open Neuroscience Journal 1, (2007).
46. George, E. L., Georges-Labouesse, E. N., Patel-King, R. S., Rayburn, H. & Hynes, R. O. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119, 1079–1091 (1993).
47. Gasiorowski, J. Z., Murphy, C. J. & Nealey, P. F. Biophysical Cues and Cell Behavior: The Big Impact of Little Things. Annu. Rev. Biomed. Eng. 15, 155–176 (2013).
48. Frantz, C., Stewart, K. M. & Weaver, V. M. The extracellular matrix at a glance. J Cell Sci 123, 4195–4200 (2010).
49. Farndale, R. W., Sixma, J. J., Barnes, M. J. & Groot, P. G. D. The role of collagen in thrombosis and hemostasis. Journal of Thrombosis and Haemostasis 2, 561–573 (2004).
50. Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006).
51. Durbeej, M. Laminins. Cell Tissue Res. 339, 259–268 (2010).
52. Dallas, S. L., Chen, Q. & Sivakumar, P. Dynamics of assembly and reorganization of extracellular matrix proteins. Curr. Top. Dev. Biol. 75, 1–24 (2006).
53. Clegg, D. O. Integrins in the development, function and dysfunction of the nervous system | Kopernio. https://kopernio.com/viewer?doi=10.2741%2F1020&token=WzEyNDAwNTEsIjEwLjI3NDEvMTAyMCJd.RgHqZza0McVG8oSr_n4tNpzb5CM.
54. Cawston, T. E. & Young, D. A. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 339, 221–235 (2010).
55. Case, L. B. et al. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 17, 880–892 (2015).
56. Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15, 786–801 (2014).
Computer Assisted Image Analysis.
Hey everyone! I am Maham Zia and this summer I worked as a post-bacc for Anna Ritz (primary advisor) and Derek Applewhite (co-advisor). I graduated from Reed in May 2020 with a degree in Physics, but during my undergraduate career I worked in a cell biology and cellular biophysics lab. I am interested in how physics interacts with other disciplines such as biology and chemistry. I believe that interdisciplinary research transcends boundaries and aids the scientific community to think about a variety of problems in creative ways.
This summer I put my coding skills to use by working on a computational project that involved analyzing images of cells. For her senior thesis, Madelyn O’ Kelley-Bangsberg ’19 used the punctate/diffuse index to measure the distribution of phosphorylated NMII Sqh. Punctate/diffuse index is widely used to measure the spread of cytochrome c in cells during apoptosis. Measuring this index involves determining standard deviation of the average brightness of the pixels using time lapse microscopy [1]. O’Kelley-Bangsberg’19 used the same idea to determine whether the cell has punctate phosphorylated myosin (high pixel intensity standard deviation) or diffuse phosphorylated myosin (low pixel intensity standard deviation). She did this in ImageJ by outlining each cell, obtaining an X-Y plot of the intensity and finally calculating the relative standard deviation ((standard deviation/mean pixel intensity) x 100 ) by setting the highest intensity value to 1 and lowest intensity value to 0. She used relative standard deviation instead of the absolute standard deviation to draw comparisons because she observed that mean pixel intensity values changed significantly between treatments [1]. Under Anna’s guidance I worked on automating this process in MATLAB by making use of image processing techniques which made the entire process a lot more time efficient.
I was working with images that each had two channels:
- An actin channel that shows the distribution of actin- a double helical polymer that aids in cell locomotion and gives the cell its shape. Since actin is distributed throughout the cell, staining it allows us to see the entire cell [2].
- A channel showing the distribution of phosphorylated NMII Sqh.
The general idea was to use the actin image to detect/outline the cells and somehow use that information to plot the cell outline on the second image which has the distribution of the phosphorylated NM II Sqh. Plotting them would allow me to obtain the intensity values of the pixels within each cell and determine the standard deviation of the pixel intensity values for each cell.
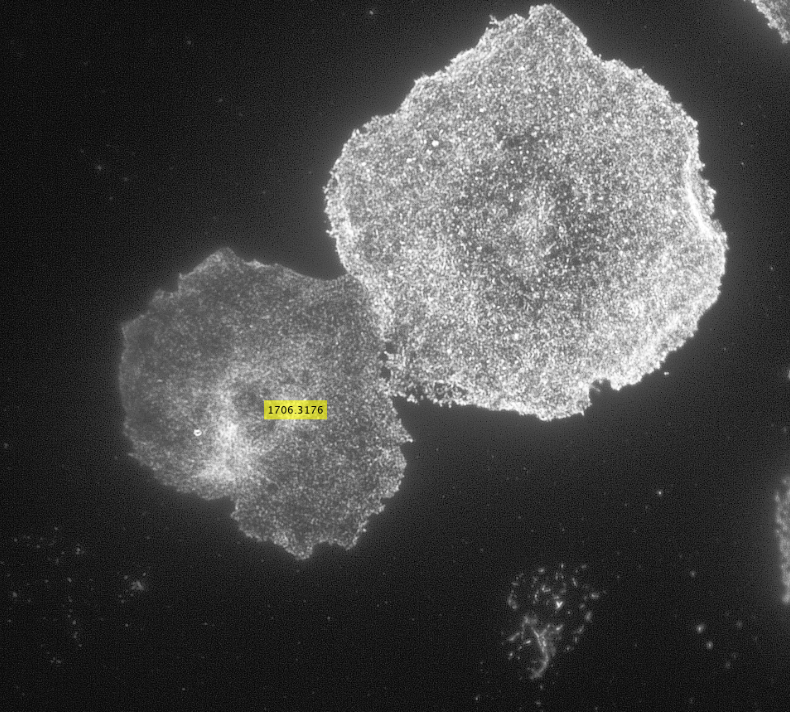
Initially, my code read in the image with the actin staining and used the inbuilt imfindcircles function in MATLAB to detect cells in the actin image. One of the input arguments of the function is radiusRange which can be used to detect circles with radii within a certain range. However, the function doesn’t work well for ranges bigger than approximately 50 pixels which means it doesn’t work well for images with both small and large cells. Moreover, the function has an internal sensitivity threshold used for detecting the cells. Sensitivity (number between 0 and 1) can be modified by putting it in as an optional argument when calling the function, but increasing it too much leads to false detections [3]. Using imfindcircles function is not a robust way to detect cells and therefore I decided to switch to the drawfreehand function in MATLAB 2020 that allows the user to interactively create a region of interest (ROI) object.

After creating the ROI object (in simple terms that means outlining the cell) as shown in the figure above, I created a binary mask and used regionprops to get the centroid and the equivalent diameter of the object.
The code then read in the second image as shown in image below with the distribution of the phosphorylated NMII Sqh and used outputs from regionprops and the impixel function to get the pixel intensity values for each cell. This image mostly looks dark because it is only showing the distribution of phosphorylated NMII Sqh which is the small cluster of bright pixels we see.
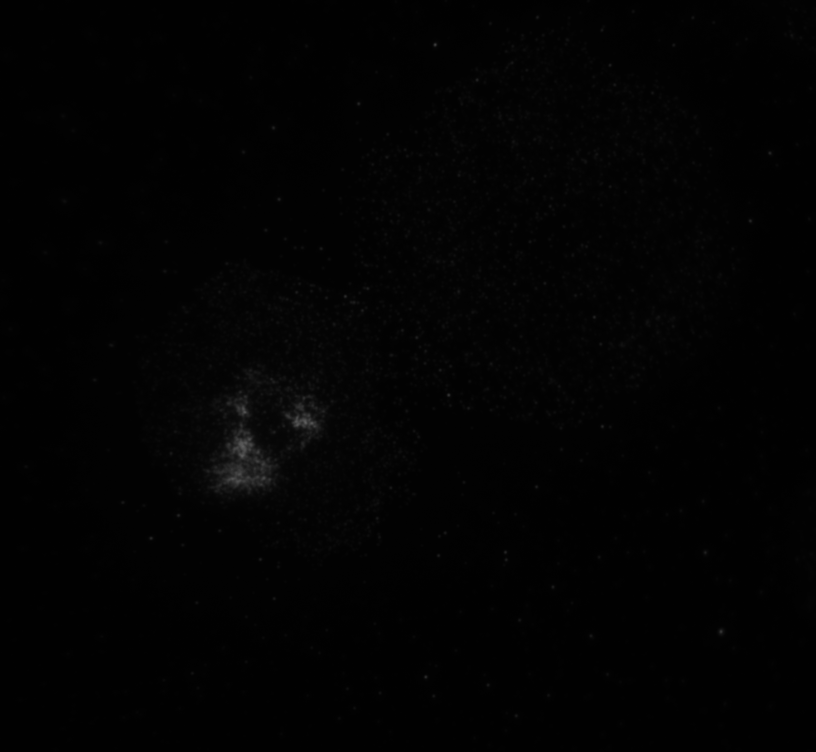
Impixel function takes the column and row indices of the pixels to be sampled and gives their pixel intensity values. However, lengths of column and row vectors need to be the same and since I was approximating the cells as circles, the only way to extract the intensity values of the pixels was to think of the circle as enclosed in a square. So, I used the center coordinates and radii of the circles to get coordinates of the top left corner of the square and used the linspace function to get equally spaced vectors for column and row. Finally, std2 and insertText functions were used for calculating the standard deviation values and displaying them on the image showing the actin distribution respectively.
Future goals could involve analyzing a number of images to determine reasonable standard deviation values and finding a way to extract pixel intensity values for pixels only within the ROI object instead of a square enclosing the object to calculate standard deviation values.
Now that I am done with this project, for this upcoming year I will be working as a research assistant in a lab part of the department of Genetics, Cell Biology, and Development at the University of Minnesota.
References:
[1] M. O’Kelley-Bangsberg, Reed undergraduate thesis (2019)
[2] J. Wilson and T. Hunt, Molecular Biology of the Cell: The Problems Book (Gar- land Science, New York, NY, 2008), 5th ed., ISBN 978-0-8153-4110-9, oCLC: 254255562.
[3] MathWorks, “Detect and Measure Circular Objects in an Image”, https://www.mathworks.com/help/images/detect-and-measure-circular-objects-in-an-image.html
Microbe Machine
Molecular cloning with Gibson Assembly
Molecular cloning is a technique established in the early 1970s that made introducing specific genes into a host possible [1]. This significantly advanced research because it allowed us to isolate and study individual genes. One method cloning has made possible is protein purification. By introducing a gene to a bacterial vector, the encoding protein is able to be overexpressed and then subjected to biochemical analyses such as crystallization or enzymatic assays to elucidate the function of the protein [2].
I am using this exact technique in my senior thesis at Reed College to study CG11811, a protein novelly indicated as a non-muscle myosin II (NMII) regulator. NMII is an actin binding protein that when activated, constricts actin filaments to change cell shape [3]. Our previous studies found that when CG11811 was depleted using RNAi, there were decreased contractility events. However, we do not know how exactly CG11811 regulates NMII. Therefore, I am cloning CG11811 into a strain of E. coli designed to mass produce proteins in order to purify it and run enzymatic assays to reveal some of its biochemical properties. The first step of this cloning process is creating the recombinant DNA plasmid to transform into the bacteria.
A very efficient technique for this purpose is Gibson Assembly, designed by Dr. Daniel Gibson in 2009 [4]. With Gibson cloning, any two sequences (the gene of interest and the vector) can be joined together without the need for restriction enzyme sites (Figure 1A). The basis of this technique is to design two sets of primers that overlap the gene and the vector (Figure 1B). When these sets of primers are used in PCR, the vector has a section at the end that is complementary to the insert and the insert has a section at the end that is complementary to the vector (Figure 1B). These two PCR products are then combined in the Gibson reaction with an exonuclease that will chew back the overlaps to create “sticky” ends that will anneal to each other, polymerase to fill in any gaps in base pairs in the overlaps, and ligase to join the fragments together (Figure 2). The resulting product is a contiguous plasmid that can then be transformed into the protein expression E. coli to get mass quantities of CG11811.
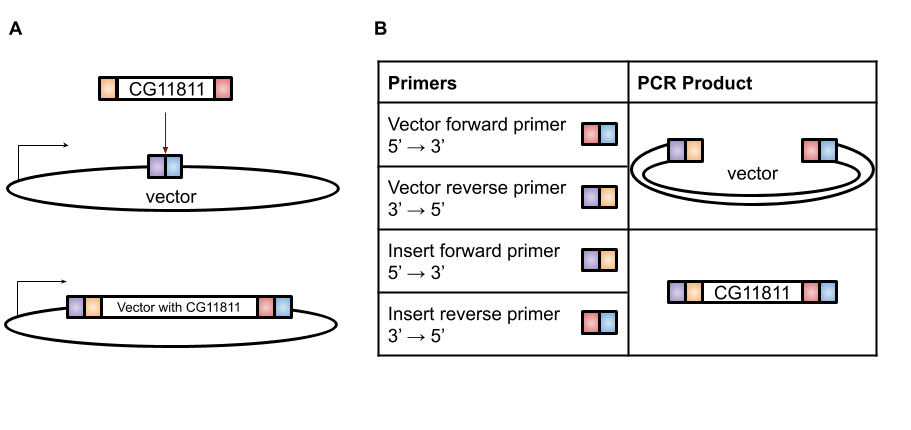

Citations
1. Jackson, D. A., Symons, R. H., & Berg, P. (1972). Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 69(10), 2904–2909. doi:10.1073/pnas.69.10.2904
2. Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Frontiers in microbiology, 5, 172. doi:10.3389/fmicb.2014.00172
3. Martin, A. C., Kaschube, M., & Wieschaus, E. F. (2009). Pulsed contractions of an actin–myosin network drive apical constriction. Nature, 457(7228), 495–499. https://doi.org/10.1038/nature07522
4. Gibson, D. G., Young, L., Chuang, R.-Y., Venter, J. C., Hutchison, C. A., & Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6(5), 343–345. https://doi.org/10.1038/nmeth.1318
Scratch-wound Assays
The scratch-wound assay is a common method for assessing protein distribution and directionality in collectively migrating cells (Cory, 2011). This method is advantageous because it does not require chemical attractants in order to induce migration. Instead, the scratch-wound assay procedure typically involves culturing a sheet of cells on top of extracellular matrix. The matrix operates like tracks that allow the cells to migrate. A needle with a fine filamentous tip is then used to etch a wound onto the sheet of tissue. The wound is then left to heal via cellular proliferation and migration, and the direction of migration can be inferred as into the wound.
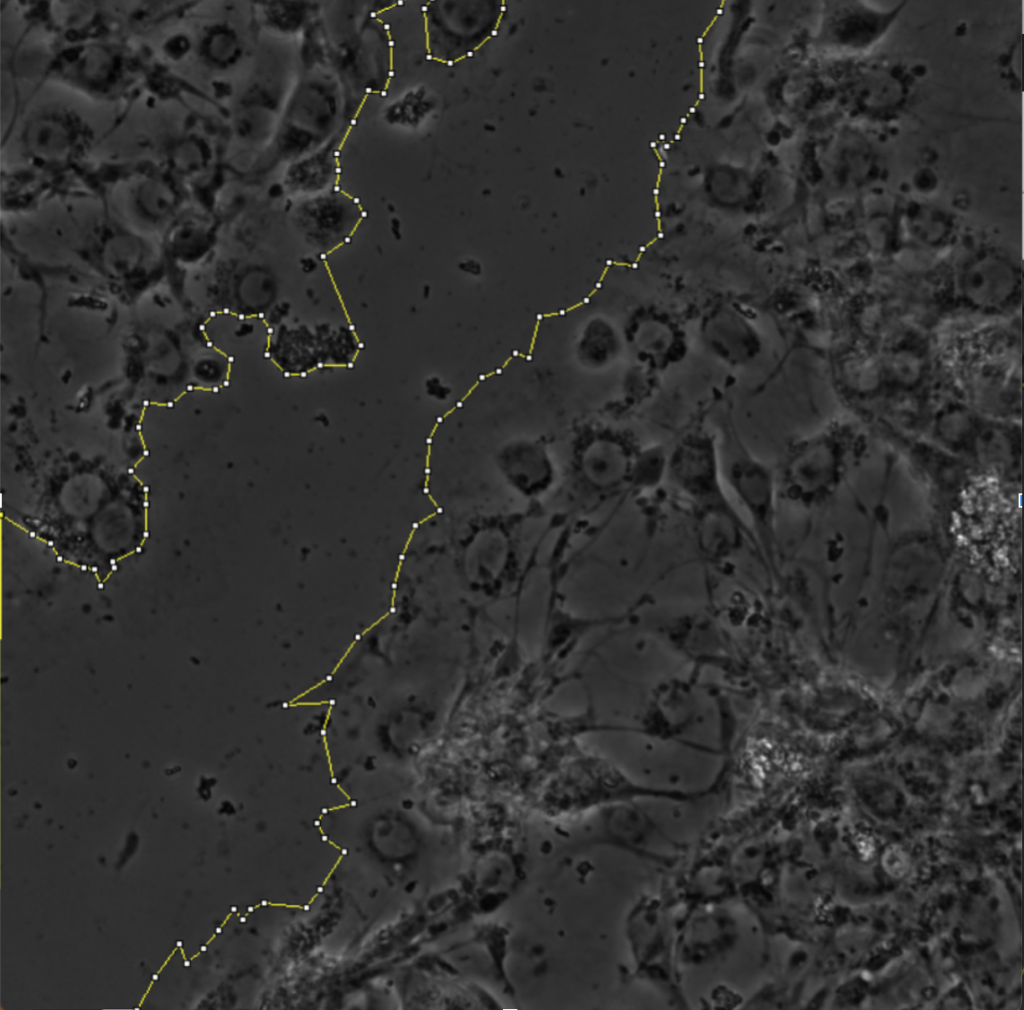
For my thesis, I will be using scratch-wound assays in order to study orientation of microtubule growth in migrating Drosophila cells. I am currently culturing sheets of sheets of RasV12;WTSRNAi cells. Some of the cultures have depleted levels of either the protein Short-stop (Shot) or Pickled eggs (Pigs), both of which crosslink actin and microtubules. Into all of these cultures, I have transfected in a DNA construct that expresses a protein called Eb1, which marks the growing ends of microtubule filaments, that is tagged with a red fluorescent molecule. After performing a scratch-wound assay, I can take three minute timelapses of the migrating cells using Total Internal Reflection Fluorescence (TIRF) microscopy. TIRF microscopy essentially uses a laser to excite the red fluorescent molecule attached to the Eb1 proteins on the growing ends of microtubules. The results are short movies in which we can see red comets marking microtubule growth. One of the goals of my thesis is to calculate the angles at which the microtubules are growing relative to the line of the wound and see if there is a difference in orientation of growth between conditions in which proteins have been depleted in cells.
References:
Cory, Giles. “Scratch-wound assay.” Cell Migration. Humana Press, 2011. 25-30.
Culturing Primary Drosophila Neurons
Though there has been great progress with understanding actin and microtubule dynamics, and their relationship to cell motility, the scientific community is still working to try to better define these interactions, and their associated proteins, that allow this movement to happen. Many neurodevelopmental disorders and degenerative diseases are linked to genes associated with the cytoskeleton (Prokop et. al., 2013), making impaired motility of neurons a possible cause. Therefore, further information on growth and motility dynamics could help lead potential treatments for these disorders.
Neurons are understood to develop an axon, their outwardly-signaling appendage, by the elongation of a neurite via microtubule recruitment. The growth cone at the tip of this developing axon is actin-rich, and forms focal adhesions with the substrate to facilitate movement and network development (Prokop et. al., 2013), making the growth cone the leading edge in neurons. Interestingly, there have been some indications that these growth cones may move at different rates on some substrates than others. Because of the substrate-dependency lead, it may be possible to utilize different substrates to characterize some of the proteins present in the extracellular matrix that contribute to efficient movement of neurons. For my thesis project I’m attempting to look at growth cone dynamics on two substrates: extracellular matrix (ECM) and concanavalin A (ConA, a plant lectin often used to plate cells) using neurons extracted from Drosophila 3rd instar larvae.
Drosophila lend themselves as an excellent model for examining the role of proteins in neuronal movement. Not only are the proteins affecting actin and microtubule dynamics well-conserved, making the findings generalizable across species, but the genetics are simplified in drosophila as well, with little redundancy in the genome. This attribute makes it so that a single knockout is capable of almost completely eliminating the function of a protein, whereas in other animals, such as mice, the knockout of multiple genes may be required to silence a protein due to redundancy (Prokop et. al., 2013). Additionally, existing knowledge about their genome makes it so that we can easily and time-efficiently make genetically altered strains for experimentation. This makes knockdown studies easy and efficacious, and desirable crosses quick to generate. Their neurons have been found to grow well in cultures, even forming networks and displaying similar electrical properties to in-vivo functioning neurons when in proper media (Küppers-Munther et. al., 2004). This allows easy visualization and analysis of neuronal movement, further contributing to their experimental value, which is why drosophila were chosen by our lab for these studies.
In a typical experiment, I extract 6-10 brains from the drosophila larvae and apply liberase to break apart the cell-cell contacts. These neurons are then plated in rich cell media on either an ECM- or a ConA-coated glass slide in a dish and allowed to attach to the bottom of the slide. After the neurons have attached, I visualize their movement by taking videos under a microscope. Additional visualization of proteins can be achieved with staining.
Sources:
Küppers-Munther, B., Letzkus, J. J., Lüer, K., Technau, G., Schmidt, H., & Prokop, A. (2004). A new culturing strategy optimises Drosophila primary cell cultures for structural and functional analyses. Dev Biol, 269(2), 459-478.
Prokop, A., Beaven, R., Qu, Y., & Sánchez-Soriano, N. (2013). Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J Cell Sci, 126(Pt 11), 2331-2341.
Actin Microtubule Crosslinking
Studies have shown that the cytoskeletal elements in the cell (actin, microtubules and intermediate filaments) engage in extensive crosstalk. This crosstalk is an important part of the regulation of the cytoskeleton, as well as a number of other biological processes. For this blog post, I will be focusing on actin-microtubule cross linking, since that is most relevant to my thesis research. Before I get into specifics about how this is related to my thesis, I’m going to give a brief, general overview of actin microtubule crosstalk and the various roles it can play within a cell.
Actin is a highly conserved protein in cells that switches between G-actin and filamentous F-actin. Actin is one of the most abundant proteins in eukaryotic cells and plays an important role in muscle contraction, cell signalling and regulation of cell shape. Microtubules are dynamic, polar and are made up of alpha & beta tubulin. The tubulin polymerizes to form microtubule filaments. Key to microtubules ability to perform their functions in the cell is dynamic instability and polarity. Dynamic instability is characterized by periods of rapid growth followed by periods of depolymerization. This allows microtubules to rapidly alter their configuration in order to fit the needs of the cell. Microtubules and actin play important roles in cell division, cell migration and other important cellular processes.
Actin Microtubule crosstalk is mainly defined through the physical mechanisms by which it occurs. This means that usually what happens is a physical linkage between parts of actin and parts of microtubules that lead to stabilization or nucleation etc. One of the main forms of actin microtubule crosstalk is crosslinking. This occurs when proteins link microtubules to actin. This linkage is enabled by large protein complexes that can also interact with microtubule plus end binding proteins. This linkage connects the plus ends of microtubules to actin bundles which can result in a redirection of microtubule growth.
One of the proteins that plays a role in actin microtubule cross linking is Short Stop (Shot). Short stop is a spectraplakin that has been shown to bind microtubules & actin filaments and has also been localized in growth cones. In my thesis, I’m looking at microtubules in Drosophila melanogaster neuroblasts, specifically at what happens when you knock down shot thus inhibiting crosslinking. I’m studying the effect of knocking down this crosslinking protein on microtubule dynamics in 3 different parts of the neuron.
Works Cited
Applewhite, D.A., Grode, K.D., Keller, D., Zadeh, A.D., Slep, K.C., and Rogers, S.L. (2010). The Spectraplakin Short Stop Is an Actin–Microtubule Cross-Linker That Contributes to Organization of the Microtubule Network. Molecular Biology of the Cell 21, 1714–1724.
Dogterom, M., and Koenderink, G.H. (2018). Actin–microtubule crosstalk in cell biology. Nature Reviews Molecular Cell Biology.
Sanchez-Soriano, N., Travis, M., Dajas-Bailador, F., Goncalves-Pimentel, C., Whitmarsh, A.J., and Prokop, A. (2009). Mouse ACF7 and Drosophila Short stop modulate filopodia formation and microtubule organisation during neuronal growth. Journal of Cell Science 122, 2534–2542.
qRT-PCR to verify RNAi knockdown
As discussed in a recent blog post, RNAi is a common technique used in the Applewhite lab to observe the effects of a silenced gene. When preparing for RNAi, it is common practice to run a sample of the dsRNA template on a gel to make sure the resulting band is the same size as the target. However, is this sufficient practice to conclude your following results are due to the knockdown of the gene? Many publishers would say no. It is possible that the exogenous dsRNA was not in appropriate concentration, or was not an effective target to cleave the specific mRNA sequences. Real time quantitative polymerase chain reaction (qRT-PCR) is a supplementary method used to verify the successful knockdown of the gene of interest.
Quantitative PCR (qPCR) is accomplished by extracting endogenous RNA from your cells treated with RNAi, reverse transcribing the RNA to DNA, designing primers to amplify the gene of interest, and using intercalating dyes, such as SYBR Green, which bind to the DNA and fluoresce with greater intensity as the concentration of the target sequence increases. Important to note, in the presence of off-target dsDNA, sequence-specific probes can be used which rely on FRET for detection, and fluoresce only when the DNA polymerase separates the quencher from the emitter. These sequence-specific probes include Taqman, Molecular Beacons, and Scorpions, although require more complex and expensive implementations (1).
The real time element is essential to determining initial DNA template concentration. Since qPCR only measures the end concentration of target sequence, there is no way to calculate an initial concentration. qRT-PCR however, measures template concentration at an exponential stage of replication, which allows for calculation of an initial starting concentration. This in turn, enables analysis of initial gene expression, and if minimal, verification of RNAi success (2).

Figure 1. Relative qRT-PCR and qPCR measurements of target concentration in respect to duration of PCR.
Sources Cited
- The Basics: RT-PCR.Thermo Fisher Scientific – USAvailable at: https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rtpcr-analysis/general-articles/rt–pcr-the-basics.html. (Accessed: 23rd October 2018)
- Bansal, R.et al.Quantitative RT-PCR Gene Evaluation and RNA Interference in the Brown Marmorated Stink Bug.Plos One11,(2016).
Mander’s Coefficient
One aspect of my thesis is exploring co-localization of Split Discs with other proteins in drosophila cells. In order to do this, not only does wet lab work need to be accomplished, but mathematical analysis (in this case using Mander’s coefficient).
Fluorescence microscopy does not have the ability to see whether or not two molecules are directly interacting. However, by looking to see if they co-localize in the cell, it can be determined whether they interact with the same complexes in the cell. The limit for fluorescence microscopy is the resolution of the images produced. Because of this, small numbers of puncta are not sufficient for determining whether or not the experimental molecules are actually co-localized. Multiple puncta from different regions within the cell must be used in analysis so the data is not limited to overlapping puncta which are a result of organelles that are close in proximity to one another.
In order to quantitatively determine the correlation of co-localization in the cell, mathematical analysis of the data is employed. For my thesis, I am employing Mander’s Overlap Coefficient (MOC) for this analysis because it does not require distinguishing fluorescence as being the result of a fluorescent protein or background noise. MOC is able to do this because it only compares the co-occurrence of fluorescence among pixels. MOC = ∑i(Ri×Gi) / √(∑iR2i×∑iG2i) where Ri and Gi are the average level of grey from the red and green fluorescence respectively (Manders et al., 1993). MOC has a range of 0 – 1 and Ri and Gi have a range of -1 – +1. The limitation to this equation is that the ratio of values can result in ambiguous numbers. Therefore, the numerator and denominator can be split up in such a way to account for the ambiguity. From this we get two coefficients: M1 (fraction of red fluorescence in areas with green fluorescence) and M2 (fraction of green fluorescence in areas with red fluorescence) (Manders et al., 1993). M1 = (∑iRi,colocal) / ∑iRi where Ri,colocal = Ri if Gi > 0 and Ri,colocal = 0 if Gi = 0 and M2 = (∑iGi,colocal) / ∑iGi where Gi,colocal = Gi if Ri > 0 and Gi,colocal = 0 if Ri = 0 (Manders et al., 1993). The larger MOC, M1, and M2 are the stronger the evidence for co-localization of the proteins within a cell. In my thesis, MOC, M1, and M2, will be gathered for each cell to determine whether or not Split Discs are co-localizing with other specific proteins.
References:
Dunn, K. W., Kamocka, M. M., & McDonald, J. H. (2011). A practical guide to evaluating colocalization in biological microscopy. American Journal of Physiology – Cell Physiology, 300(4), C723–C742. http://doi.org/10.1152/ajpcell.00462.2010
Manders, E. M., Verbeek, F. J. & Aten, J. A. (1993). Measurement of co‐localization of objects in dual‐colour confocal images. Journal of Microscopy, 169, 375-382. doi:10.1111/j.1365-2818.1993.tb03313.x