When attempting to understand a protein’s function in a cell, the effects of removing that protein can be very telling. As such, knockdowns and knockouts, techniques which remove a targeted protein from acting in the cell, are widely used to identify how a cell behaves without the functions performed by said target protein.
A knockout is an irreversible procedure that removes the target protein from the cell permanently, often by editing the genome itself to either deactivate or directly remove the target protein’s coding sequence. This can be achieved by selective breeding when dealing with whole organisms, but when working with cell cultures, knockouts are typically accomplished by means such as the gene-editing kit TALEN or, in recent years, by use of the tool CRISPR-cas9.
A knockdown, on the other hand, is a repeated procedure which removes the target protein from the cell temporarily. By treating cells on a regular basis, the target protein is disrupted from its usual function, and the cells can be observed without its influence. If the treatment ceases, the target protein will no longer be disrupted and the cell will return to normal function, if mildly worse for wear. The distinction between these two procedures can be likened to ending a fight (knockout) as opposed to temporarily ‘gaining advantage’ or suspending the fight (knockdown).
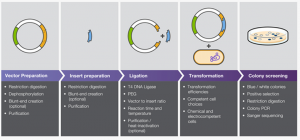
In our experiment, we have performed a RNA interference (RNAi) knockdown of our target proteins flw and CG. This is achieved first by the transformation of genomic DNA (gDNA) that codes for the target protein into double stranded RNA (dsRNA), and then by simply exposing cultured cells to the dsRNA on a sustained and regular basis.
The reason this apparently simple method (which requires more pipetting and tube shuffling than that short sentence might imply) works and removes the target protein from action is due to the cell’s own inherent defensive mechanisms. When exposed to free floating dsRNA in solution, some dsRNA is naturally taken up into the cell, where it is recognized as foreign and chopped to pieces. Ironically, this causes the fragmented dsRNA to bind to messenger RNA (mRNA) already in the cell that matches its sequence, whereupon the entire dsRNA-fragment-mRNA amalgamation is recognized as foreign and chopped to pieces.

Figure 1. dsRNA suspended in cell media is taken into a cell, recognized as a foreign component, and cut into pieces by the appropriate enzymes. The cut pieces of dsRNA attach to pieces of mRNA naturally present in the cell, which are subsequently tagged for sheering due to their binding with a foreign component.
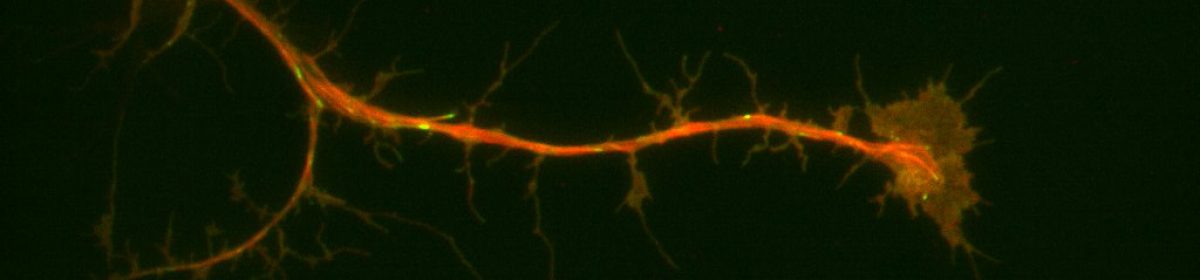
When the dsRNA is in sufficient concentration, this defensive response results in the cell being unable to translate the appropriate mRNA into the target protein, since it is instead destroying that mRNA as fast as it can. Once depleted of the target protein, cells can be treated, fixed, stained, or any combination thereof, and the effects of the knockdown can be observed to extrapolate the target protein’s function.