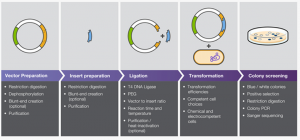
BirA is biotin protein ligase in E. coli that selectively adds biotin to a subunit of acetyl-CoA carboxylase. Roux et al. created a mutant BirA that isn’t selective to its native substrate and will instead biotinylate any proteins that are close-by. This allows for identification of protein-protein interactions in eukaryotic cells by creating a fusion protein of BirA and a protein of interest that will biotinylate proximal proteins, which can then be captured and identified. Protein identification utilizes the strong association of biotin and streptavidin (or avidin) to capture proteins that have been biotinylated by BirA, followed by mass spectroscopy to identify biotinylated proteins.
Figure 1. Schematic of BioID assay from Roux et al. 2012.

I am investigating the protein interactions of an actin-microtubule crosslinking protein coded by the gene SPECC1L (with an ortholog known as “split discs” in Drosophila). Mutations in SPECC1L have been identified in a number of diverse cases of orofacial cleft, and thus knowledge of protein-protein interactions by the product of SPECC1L is particularly important to understanding the developmental basis of this set of conditions.
To this end, I am using a BirA-split discs construct to investigate what proteins split discs interacts with in vivo in Drosophila. This construct will ideally biotinylate proteins that split discs typically interacts with, without excessive non-target biotinylation and without affecting the behavior or split discs. I will then capture biotinylated proteins using the biotin-streptavidin interaction and identify using mass spectrometry.
References:
Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. The Journal of Cell Biology. 2012;196(6):801.