
NMR & IR Tables
1H NMR Chemical Shifts
Commonly observed 1H chemical shifts fall in a narrow 14 ppm range. However, chemical shifts associated with particular structural units fall in even narrow ranges as shown by the following chart.
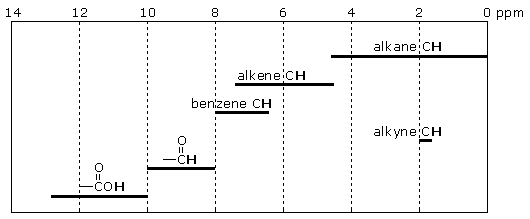
Some structural units, such as alkenes and benzene rings,produce signals in the same chemical shift range. Therefore, you cannotrely entirely on chemical shift values to identify these structural units.
You may have noticed that the structural units in the chart are limited to CH-containing units (the sole exception is -CO2H). OH and NH units produce signals within the 0-14 ppm range, but the positions of these signals are strongly influenced by hydrogen bonding and sample concentration, and do not reliably correlate with chemical structure.
Chemists have found that useful chemical shift-structure correlations also exist within the alkane, alkene, and benzene CH families. Some of these correlations are described below.
Alkane CH. The chemical shift of an alkane CH depends on the other groups bonded to carbon. The following table lists typicalchemical shifts for methyl hydrogens, that is, for XCH3groups. (The table has been adapted from a reserve text: Pretsch etal., Tables of Spectral Data for Structure Determinatio nof Organic Compounds, Springer-Verlag, Berlin, 1983, p. H5.)
| XCH3 | Typical Chemical Shift (ppm) |
|---|---|
| R3Si-CH3 | 0 |
| R-CH3 | 0.9 |
| R2C=CR-CH3 | 1.7 |
| RCºC-CH3 | 1.8 |
| NºC-CH3 | 2.0 |
| RC(=O)-CH3 | 2.1 |
| I-CH3 | 2.2 |
| Ph-CH3 | 2.3 |
| R2N-CH3 | 2.5 |
| Br-CH3 | 2.7 |
| RC(=O)NR-CH3 | 2.7 |
| Cl-CH3 | 3.1 |
| RO-CH3 | 3.2 |
| RC(=O)O-CH3 | 3.7 |
| PhO-CH3 | 3.7 |
| F-CH3 | 4.3 |
| Add 0.4 if -CH2R | Add 0.8 if -CHR2 |
Most XCH3 signals appear at larger chemical shifts than RCH3. Chemists describe this behavior in two ways. We say, “X deshields the methyl hydrogens.” Or, we say, “X shifts the methyl signals downfield” or “X shifts the methyl signals to lower field.” The origin and meaning of these phrases can be found in various textbooks (see the Chem 201/202 Reserve book list) and won’t be delved into here.
An X group can produce deshielding effects in multiple ways. The deshielding effect of X is roughly correlated with its electronegativity. The chemical shifts of XCH3increase X = C < X = N < X = O < X = F. Unsaturated groups, like X = alkene, benzene, and carbonyl, also exert a deshielding effect (relative to X = R).
As you use this table of chemical shifts, keep in mind that these values are “typical.” XCH3 shifts can easily vary by as much as 0.5 ppm from the values listed here.
Another limitation of this table is that it does not provide shifts for methylene (XCH2Y) or methine (XYZCH)hydrogens. The chemical shifts of these hydrogens reflect the combined (de)shielding effects of X, Y, and Z, however, one cannot simply add the effects of these groups together.
The effect of multiple deshielding groups can be seen in the spectrum of the cyclic acetal shown below. Every –CH2-group is attached to at least one O, but the methylene group with two O substituents is far more deshielded (4.9 ppm) than the groups with only one O substituent (3.9 ppm).
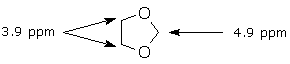
If Y and Z happen to be alkyl groups (R), it is easy to predict crude chemical shifts for the methylene and methine hydrogens. The chemical shift of the methylene hydrogens in XCH2R is usually 0.4 ppm greater than the chemical shift of XCH3. Similarly, the chemical shift of the methine hydrogen in XCHR2 is about 0.8 ppm greater than the chemical shift of XCH3.
For example, the methylene protons in ethyl acetate, CH3C(=O)OCH2CH3,are predicted to have a shift of approximately 4.1 ppm (3.7 for C(=O)OCH3+ 0.4 for a methylene group). The experimental value is 4.0 ppm, so the prediction is not too bad.
Alkene CH. The chemical shifts of alkene hydrogens are affected by other groups attached to the two alkene carbons. Each group’s effect depends on its spatial relationship (geminal, cis,or trans) to the hydrogen of interest, and the combined effects of different groups are added together as shown below. (The following table and formula for predicting alkene shifts have been adapted from a reserve text: Silverstein et al., Spectrometric Identification of Organic Compounds,5th Ed., Wiley, 1991, pg. 215.)
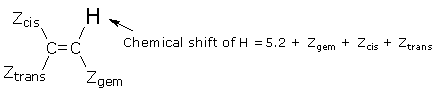
| Substituent Constants for Alkene H Chemical Shifts | |||
|---|---|---|---|
| Substituent | Zgem | Zcis | Ztrans |
| -H | 0 ppm | 0 ppm | 0 ppm |
| -R | 0.6 | -0.3 | -0.3 |
| -C=C | 1.0 | 0 | -0.2 |
| -CºC | 0.5 | 0.4 | 0.1 |
| -CºN | 0.2 | 0.8 | 0.6 |
| -Ph | 1.4 | 0.4 | -0.1 |
| >C=O | 1.1 | 1.1 | 0.8 |
| -SR | 1.0 | -0.2 | 0 |
| -Cl | 1.0 | 0.2 | 0 |
| -Br | 1.0 | 0.4 | 0.6 |
| -NR2 (R = alkyl) | 0.7 | -1.2 | -1.3 |
| -OR (R = alkyl) | 1.2 | -1.1 | -1.3 |
The following example shows how chemical shifts are calculated for the two alkene protons in cyclohexenone. One starts with a “base”of 5.2 ppm and then adds the (de)shielding effects of other non-hydrogen groups depending on their structure and location.
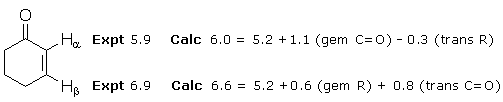
Benzene CH. Hydrogens attached to benzene rings give distinctive signals between 6.7-8 ppm. The signals tend to be large because there are usually four or five hydrogens attached to the ring. (Alkene hydrogens, on the other hand, usually create much smaller signals because there are usually only one or two of them.)
Ring hydrogens typically produce signals near 7.2 ppm, and most ring substituents have only a modest effect on the chemical shift sof the ring hydrogens. Certain groups, however, can produce significant (de)shielding effects depending on their ring position and electronic character.
The largest (de)shielding effects are due to the following groups:
- Electron-donating groups (-NR2, -OR). These groups feed electron density into the ring and shield orthohydrogens. Typical shifts for ortho hydrogens are 6.4-6.8 ppm.
- Unsaturated electron-withdrawing groups (-NO2,>C=O). These groups remove electron density from the ring and deshield ortho hydrogens. Typical shifts forortho hydrogens are 7.8-8.2 ppm.