
Most of what we are doing is not computational. We have been creating RNAi to inhibit our genes of interest. It’s tricky with drosophila: only a handful of the candidate genes are expressed in our cell line and only a subsection of that was capable of RNAi production. Despite this, we managed to produce 4 RNAi lines for these genes.
Category: Collaborative REU
Week 9: Cell tracking
So now we are in the experimental validation phase. This week, we ran a trial run of the cell tracking software, using newly cultured cells from a drosophila melanogaster, or fruit fly, line. These lines are suitable for validating that the genes are involved in cell motility due to the high degree of conservation between humans and fruit flies in basic cellular mechanisms.
Summer Week 8: Positive and Negative Sets
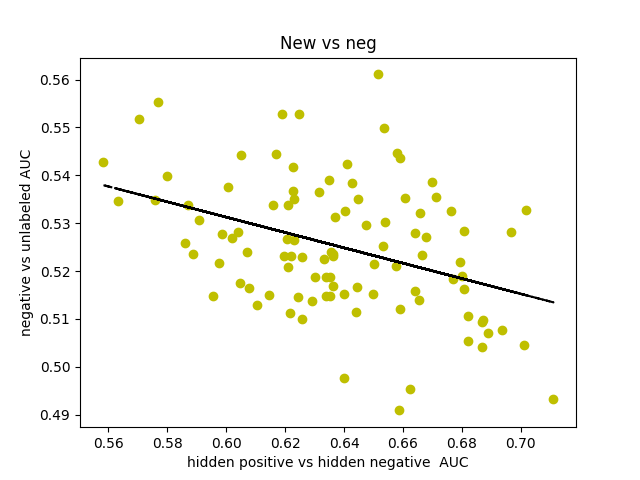
I was curious about what our AUC would look like if instead of comparing hidden positives to unlabeled, we compared hidden positives to hidden negatives, wondering if that would get us a more valid representation of the network’s ability to predict genes associated with schizophrenia. What if the unlabeled genes ranked above the hidden positives were simply actual centers of genetic perturbations in schizophrenia?
Oddly enough, the scores actually decreased from 0.65-0.70 to 0.60-0.65. I found that the AUCs for comparing hidden negatives to unlabeled nodes hovered around 0.53, meaning that the hidden negatives were ranked mostly in the top half of the list.
This somewhat makes sense though. The basis for the negative set is that they are genes in non-neurological diseases, but that doesn’t necessarily exclude them from being included in a more subtle, polygenic disorder that’s dependent on additive effects of hundreds of genetic perturbations. It also makes sense that genes involved in other diseases might have a slightly higher probability of being associated with some other disease.
I decided to make a new negative set. I started by gathering every single node in the 0.150 network. There is a resource called SZGR 2.0 (https://bioinfo.uth.edu/SZGR/) that collects all sorts of evidence for genes being associated with schizophrenia. Using this list, I excluded any gene that had any evidence for being associated with schizophrenia. I then took differential gene expression data from autism spectrum disorder, bipolar disorder, and major depressive disorder as well as schizophrenia (http://science.sciencemag.org/content/359/6376/693). If a gene was not differentially expressed in any of these diseases (FDR>0.5 for schizophrenia, FDR>0.2 for others) and was in the other list, I added that gene to my list of negatives, which was 1561 genes long. I found that this list, when hidden negatives were compared to unlabeled genes, had an AUC of about 0.44, which means that our negatives are finally oriented around schizophrenia rather than non-neurological diseases. I also found that the AUC for hidden positives vs hidden negatives spiked up to 0.75, meaning that our program is legitimately good at predicting genes associated with schizophrenia.
I found no significant performance differences from factoring in evidence levels.
Summer Week 7: Finalizations
Changed our SZ positives to be evidence based. The number of layers the positives are in is determined by the evidence level. It does better than before. We are confident in the gene lists and are looking into the candidate genes for experimental validation
Week 23
I ran the bulk-BLASTer to find good homology targets from our results
Derek Applewhite, our cell biology advisor, noted that several top genes could be novel to flies, and several genes could be novel to cell motility.
Miriam and I will continue to work out the bugs in the verification system for our process.
Week 23
This week, my goals have been relatively simple and short. There are a couple of bugs in my code that I need to sort out. I haven’t been able to collect any data about how “good” our method is due to these bugs, but once I do, we will hopefully be able to see that the results we are getting are not random.
Week 22
Since the last time I wrote a blog post, I have mainly been working on one way of verifying our algorithm; essentially, we need to make sure that our method is actually good at doing what it’s supposed to be doing. Although there are several ways to verify an algorithm, some of which Alex has been working on, I am working on something called “k-fold cross validation.”
This method of verification works by removing (or “hiding”) 1/k-th of your positives, then running your method and seeing where the hidden positives are ranked. You randomly choose the 1/k positives to hide and do this several times. You can also use the distribution of scores you get from each run to see how your method (with all positives) compares – are you getting significant results or are you getting what is expected from randomly choosing positives?
My first attempt at this was just a test run, and there were a few mistakes that I made. First, I chose to sample half instead of choosing a more reasonable number of positives such as 1/4 or 1/5. I also didn’t randomly choose positives to hide – I simply deleted the second half of the positives, and in doing so, might have removed an entire cell motility pathway or two from the positives. This would have produced biased results.
This coming week, my goal is to randomly sample 1/4 of positives multiple times, as well as implement some plotting functions in order to visualize the distribution of scores.
Week 22
So we have a BLASTer to see which genes in humans are homologous to genes in drosophila melanogaster, our model organism for cell motility. It works- it just takes a couple of hours to get an output file for 20 genes. Should be no problem to run overnight.
Miriam has been working on a way to verify that the network is working. Currently, she has been eliminating some positives while keeping others, looking at how high up the eliminated positives are in the result. It should be fruitful.
We also looked at how other functionally similar genes we were certain about were similar. We found that all of the gene interactions we thought of as functionally similar had strong edges in the networks. This validates the network as a whole as biologically sound and will further validate our results.
Week 21
Although we had the bio qual, we still managed to do a little bit of work. One of the sources for our positive set was a little suspect, so we decided to take it out and see the impact on the results. We found that there was a drastic change. Since we were relatively confident that the other positives were good, if the suspect one was good, then the results wouldn’t change as much if we ran the whole network through. What we found was that it was indeed not very good. The output file was dramatically different. But we need a way to quantify the change
This upcoming week, we will also be developing a program to find how conserved our output genes are in fruit flies, which are our model organism
No Post
I am taking the biology junior qualifying exam this weekend, so there will be no blog post. The junior qualifying exam is a cumulative exam each student must take their junior year for their major. Each student must pass it in order to be able to move on to writing a thesis their senior year and graduate.