Last week, we were lucky enough to attend the GCC/BOSC conference hosted at Reed. Although I spent the majority of the week volunteering and attending sessions, we also had a deadline for a two-page extended abstract of our CNB-MAC paper to be submitted to the main conference proceedings. We successfully cut down our original paper into two pages and submitted the abstract on Friday.
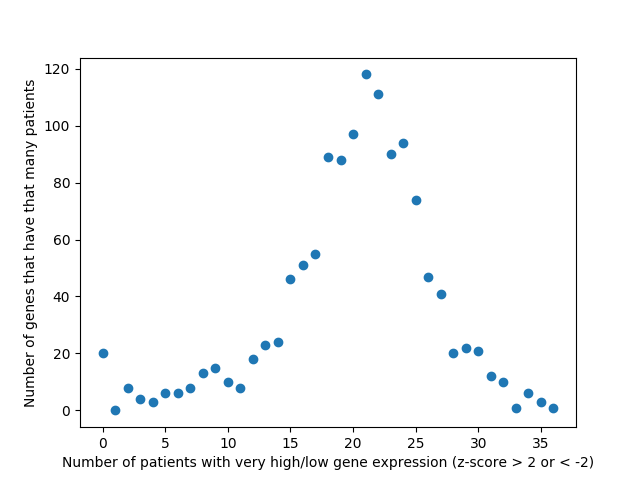
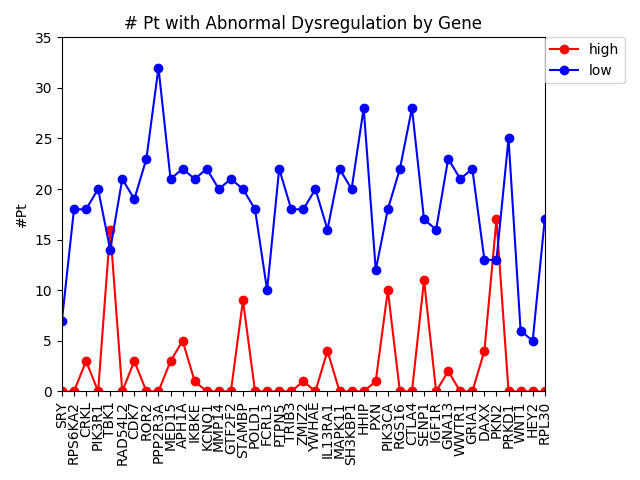
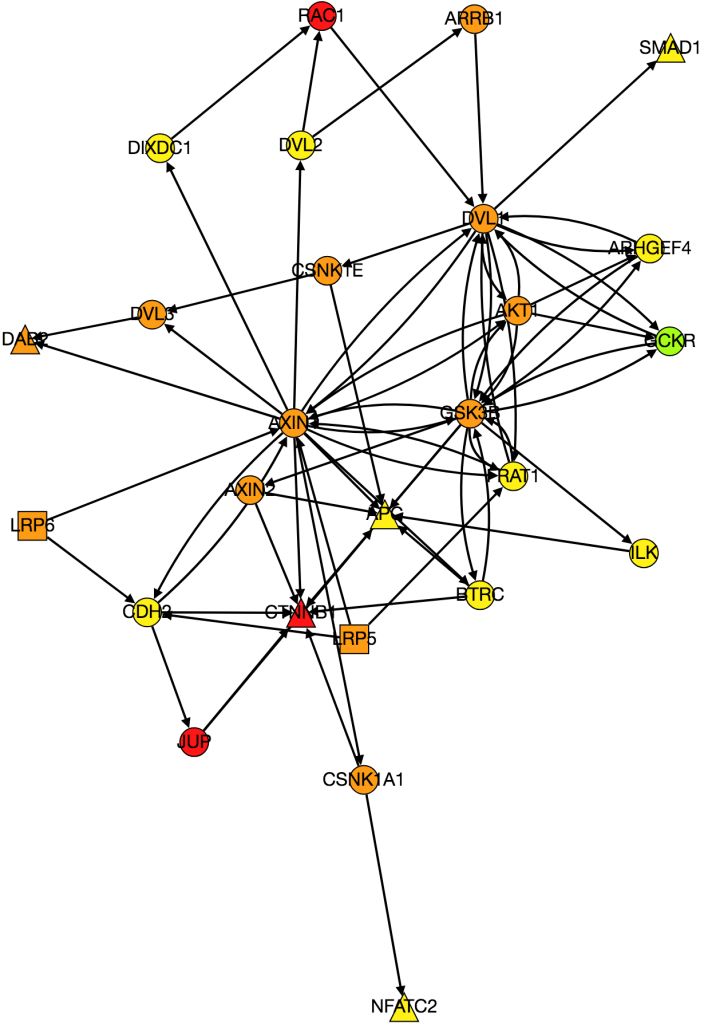
As of now, we’re almost ready to dig into the experimental portion of this project, but there are still a few things we need to iron out. At the end of last week, Alex noticed a “bug” in how we’ve been calculating the AUC of our multi-layer algorithm. Therefore, one of our priorities this week is to find a more accurate AUC calculation that better represents our method. Once that is completed, we will move on to assembling a list of candidates for experimentally validation. Hopefully by the end of the week we will have chosen 8-10 candidates that are not only biologically interesting but conserved in Drosophila as well.