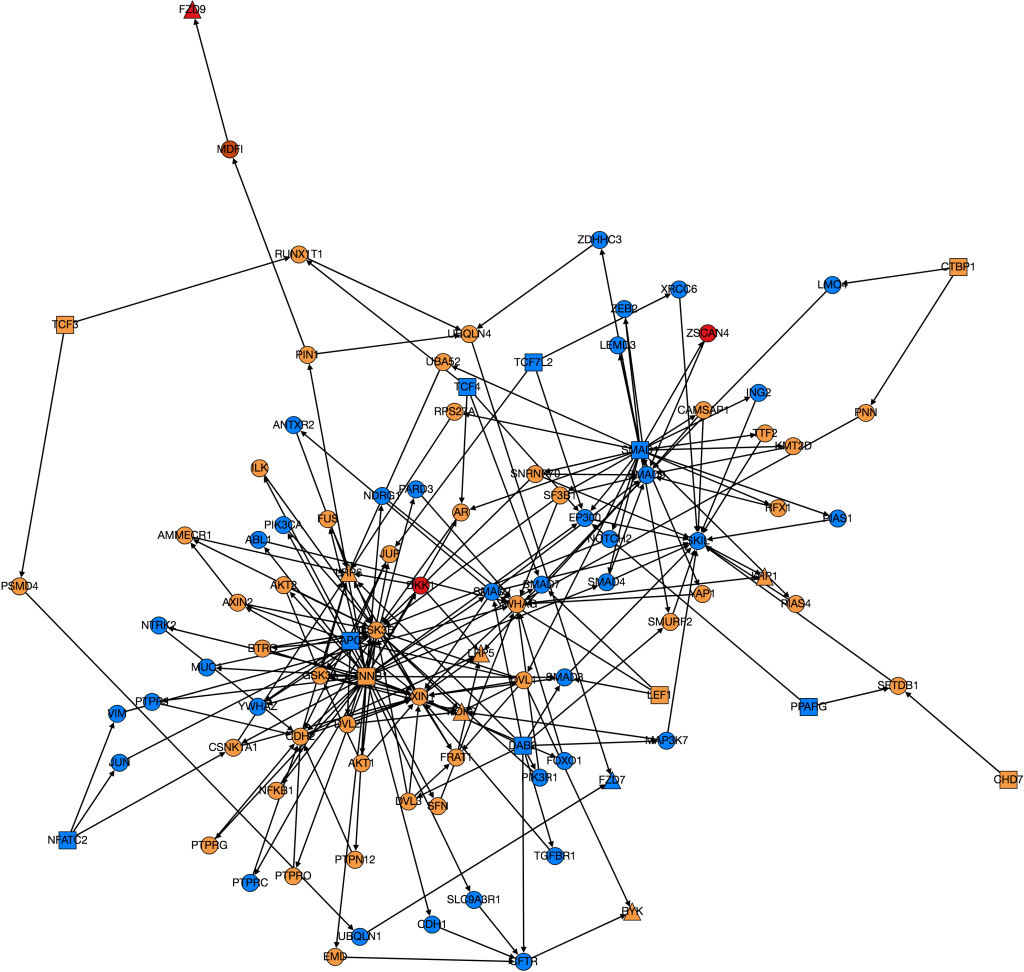
I finally managed to download all of the GO Terms and spent some time processing the data and editing previous code so that it all fit together. After having done so and obtained a working version of the code, an initial run revealed that the run time of weighting the entire HIPPIE Interactome would be approximately 101 days. This is outside the duration of the CREU so clearly something needed to be done. To fully explain the issues that lead to such a terrible run-time it is important to first have a rough understanding of the structure of the code.
This iteration of the code first produced a list of true positives and a list of true negatives on the criteria of GO Term co-annotation. This section of the code takes about 4 minutes (which can also be potentially sped up later on) but this negligibly contributes to the 3 month run time. The next portion implements functions that in conjunction calculate the cost of a given node. The necessary inputs are
- A list of all the evidence types.
- A dictionary where the keys are the evidence type labels and the values are lists (evidence lists) whose elements are the protein edges (represented as lists) which are confirmed to occur by that particular evidence type
- A list of true positives and a list of true negatives.
Finding the cost involves calculating the intersection and symmetric difference of the positives with the evidence lists and then the intersection and symmetric difference of the negatives with the evidence lists. This was done each for each evidence list “every” time a new edge was weighted. This approach took roughly 2.5+ minutes per edge. With 93138 edges, this was the main contributor to the 3 month run time. Further inspection reveals that the time-complexity of performing these operations on a set is O(min(len(s), len(t)) and O(max(len(s), len(t)) for a list. Performing this operation every single time was therefore very costly. Simply calculating these numbers for each edge at the outset and then appending them in a particular order to the end of each evidence list reduced the time needed to weight each edge down to approximately 0.45 seconds thus shaving down the total run-time to about 11 hours.
Another change that will significantly reduce the run time further is using sets instead of lists for the protein edges in the dictionary and representing the edges themselves as tuples. This will mean creating a different dictionary with identical keys but the values will the four calculated values mentioned above. This will supposedly shave off a large chunk of time because element look up in a set is significantly faster than the same operation in a list. Another idea is to change the order of calculations in a way that reduces the amount of time the exact same calculation is executed or to calculate the edges in a way that iterates over the approximately 230 evidence types rather than the 93138 edges.