
This week, there is a conference on campus for Galaxy and open source bioinformatics projects. Also, we’re refining the code.
Category: Uncategorized
Summer Week 4: Vacation
I was on vacation
Summer Week 3: Figures and papers
This week, we scrambled to make figures for the paper we are submitting to the conference. Mostly just writing and figure making.
Week 4: Cleaning up
With our big CNB-MAC paper deadline passed last Friday, my tasks this week have been relatively simple. My first goal this week was to take the bar chart figures in the paper and turn them into box and whisker plots.
For example, I took this figure:
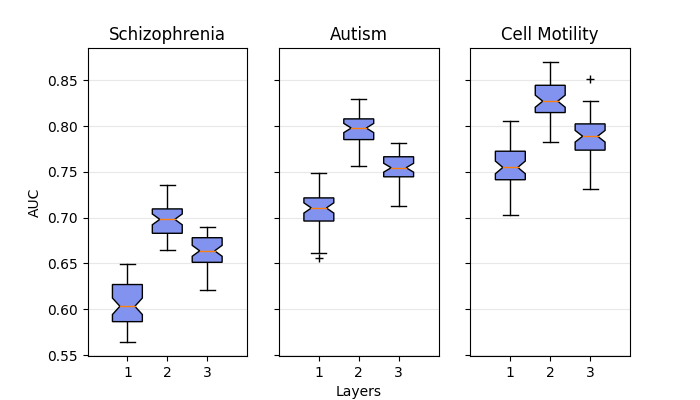
and turned it into this:
The bar chart figure shows the mean AUC of each positive set with varying number of layers, while my figure is a little more descriptive. It shows the mean AUC as well as the interquartile range and outliers.
Next, we got some surprising news! We were notified that after a preliminary pass of the paper submissions, our paper was accepted to the CNB-MAC conference as either a talk or a poster (we’re still waiting to hear which one.) This gave us the opportunity to submit a two-page abstract by the end of next week, a more challenging task. I wrote up a quick rough draft that I will continue working on next week with Alex.
Finally, with our results generated and our paper submitted, it was time to clean up our github repo. This included deleting a lot of old code and useless output files as well as restructuring the code and writing README.md files. It’s looking a lot better now!
Week 5: Visualizing Data
This week I have continued trying to visualize and integrate gene expression data from GDC TCGA onto nodes in Graphspace. This is the graph I have so far:
The color of the nodes on the graph represent the mean foldchange of the gene. The foldchange is the ratio of gene expression of a cancerous tissue sample to a normal tissue sample from the same patient. There were 41 patients with colon adenocarcinoma who had gene expression data from both a tumor and a normal sample. The foldchange for each of these 41 patients was averaged to find the mean foldchange of each gene. Light blue indicates that the gene is slightly underexpressed in cancerous samples (less than 1 standard deviation), dark blue(not shown) indicates that the gene is underexpressed by 2 standard deviations, purple (not shown) indicates that the gene is underexpressed by greater than 2 standard deviations. Orange indicates that the gene is slightly overexpressed (within one standard deviation), dark orange indicates that the gene is overexpressed by 2 standard deviations, and red indicates that the gene is overexpressed by greater than 2 standard deviations.
One problem that came up was that some genes have a lot more variance in patient data than others. (Data on variance is available by clicking on the nodes in Graphspace). Additionally, patients that no gene expression or close to no gene expression in their normal samples had extremely high foldchanges that threw off the mean. It’s tempting to just throw away those samples as outliers, however several samples had multiple “outliers”. As an example I’ve included three sets of line graphs I made that show the gene expression data and foldchange of each patient for three different genes:
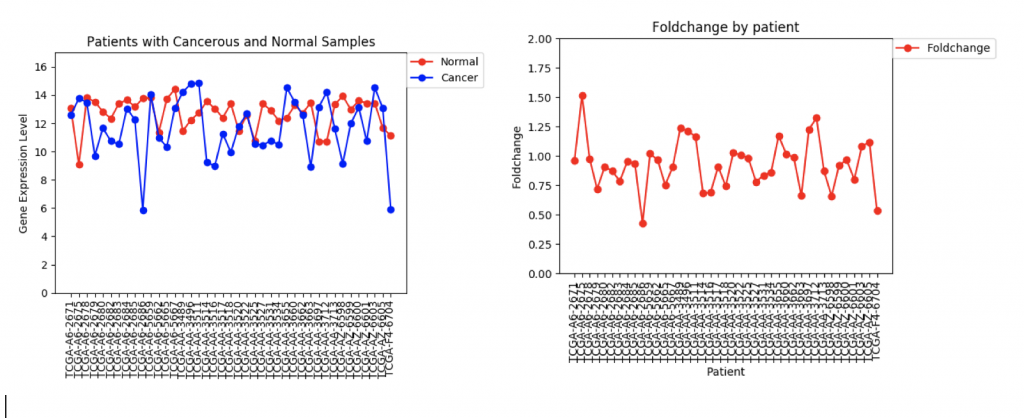
In the first example, CFTR, one can see that cancerous samples tend to have lower gene regulation than healthy samples and most of the fold change values hover around 1.0, meaning that the change is fairly low.
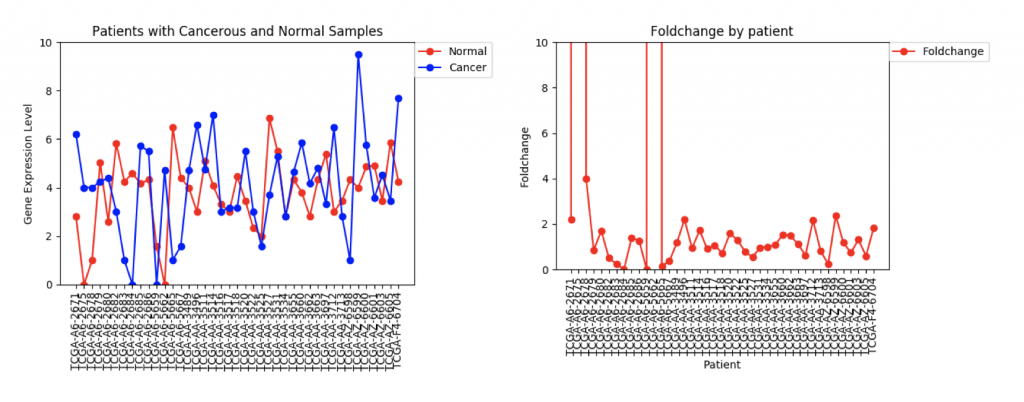
The second example, FZD9, is slightly different. The FZD9 expression data is much less uniform. In some patients, FZD9 is largely overregulated in cancerous samples. In other, it’s largely under regulated. The foldchange data shows foldchanges that range from zero to extremely high values (greater than 30,0000). This occurs because those patients had normal sample values of 0.0. In this case, it looks like dismissing the two extremely high foldchanges as outliers would yield a more realistic data set.
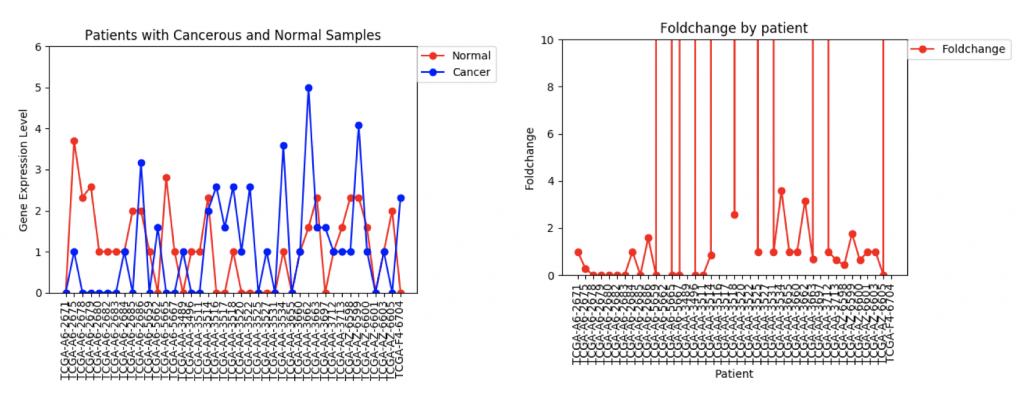
However, in ZSCAN4, there are many patients with extremely high or low foldchanges. Instead of ignoring these patients, I think I need to find a way to normalize the data so that a few large foldchanges don’t completely throw off the data.
The next step in this project is to work with Usman and Kathy to integrate the gene expression data onto the edge weights so that PathLinker can calculate the most disregulated protein pathways. To begin this process, I’ve written a program that produces a text file that contains for each gene the ID, common name, mean foldchange, standard deviation, and either a +1, 0, or -1, which represents whether it’s under-regulated, unchanged, or over-regulated in cancerous samples. Right now the +1, 0, and -1 are really just placeholder values. I need to meet with Ibrahim and Anna to discuss how to better weight the nodes.
Week 4: Obtaining and Processing Data
This week our main goal has been to find a pipeline to obtain TCGA data in a neat form. We discovered UCSC’s Xena Browser, which has files from the TCGA and a number of other databases.
Last week, we used the data from FireBrowse to make a graph of the genes that have patients with abnormally high or low levels of expression.
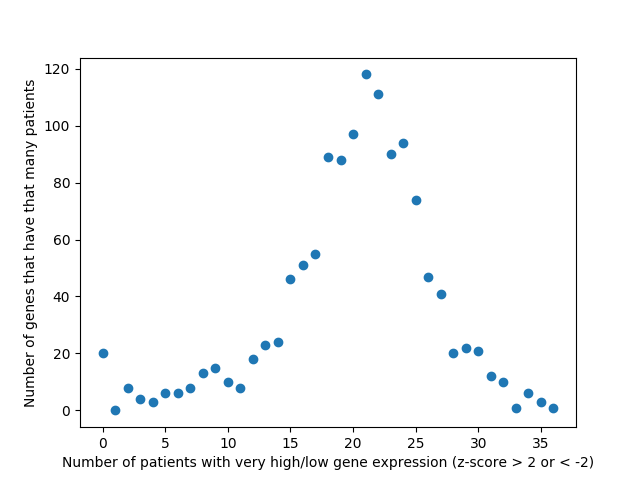
This week we changed that graph slightly by showing the difference between the number of patients with high expression and the number of patients with low expression by gene.
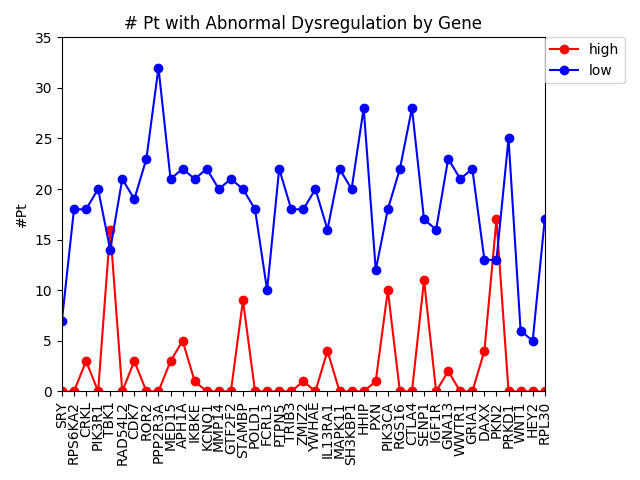
It is interesting to me that there are generally more patients with severe under-expression rather than severe-overexpression. I wonder if this is because these genes play a role in suppressing tumors, and that therefore maybe under-expression is more likely to cause cancer than overexpression?
I also worked on integrating gene expression data from Xena into our graph of the Wnt pathway.
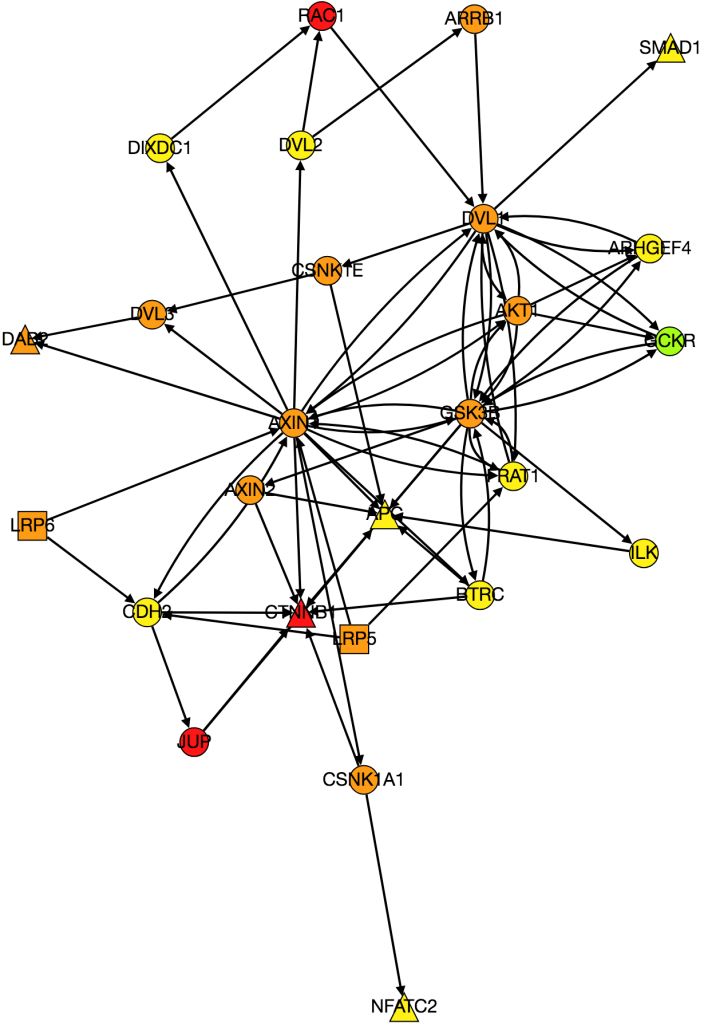
Kathy figured out what was wrong with PathLinker the first time we ran it and re-ran it. I am working on turning it into a graph, but the input data is very different because it’s coming from a different version of NetPath so I need to change the program to be able to process the new data.
I also noticed while I was processing the expression data from Xena that there was a large amount of variability in gene expression between patients. I’m currently working on several things. Instead of just averaging gene expression for genes I’m comparing gene expression patient-by-patient so I’m comparing a tumor sample to a normal tissue sample for every patient. I also want to come up with a way to visualize the variance of expression among patients, because the more variance there is the less significant differences in expression between cancerous and normal tissue are. Anna suggested I do this by making the borders on nodes with high variance thicker. I am also going back and checking my math on gene expression to make sure that it is actually statistically significant and is conducted in a way that is similar to how other researchers have done similar research in the past.
Week 3: Exploring patient data
This week, we explored the patient data from the cancer genome atlas project using firebrowse. I used the fbget tool to download 1260 files relating to colorectal cancer, each with different genes and data relating to gene expression. The figure below shows the different gene expressions.

Week 3 – Implementing Bayesian Weighting Schemes
The achievements from last week as well as the goals were laid out in this progress report. Accordingly, I spent the majority of this week working on an implementation of the Bayesian Weighting Scheme I discussed last week. This involved carefully fleshing out all the minutiae of the relevant paper (which I laid out and explained in this document from last week). I then applied the code onto a small toy-network that I made myself and then hand-calculated a few edges to verify my implementation.

I am currently working to implement the weighting scheme onto the HIPPIE Interactome. Thus far this has meant simply writing code to read the relevant text files and format the data efficiently in a way that meshes well with code I wrote for the toy example. Eventually, I will compare the scores of edges from this weighting scheme to the ones on the actual HIPPIE Interactome and further analysis on the differences between the two weighting schemes will probably be among the next steps. Besides implementing the Bayesian Weighting Scheme I have also been working to understand the underlying statistics behind the methods used to weight the HIPPIE Interactome. I will try to have a document that explains the mathematics for this in similar fashion to the one for the Bayesian Weighting Scheme.
Week 2: Sinksource+ and AUCs
We found an algorithm that solves the edge case problem. Sinksource+ removes the negatives and adds one negative to every single node. The weight of the edge is also a constant, which means that it’s the same for a node of any degree. This significantly devalues nodes that aren’t very well connected and would not be suitable for polygenic analysis.
However, it works horrendously in a single layer method with no prime nodes. The AUC sticks at around 0.5, which is the worst possible AUC you can have. However, it works great in a two layer network, giving our SZ network a score of 0.65. If you add negatives back in with the addition of sinksource+ and reduce the sinksource+ constant, we get an SZ AUC of 0.675 and a cell motility AUC of 0.801, which is the best AUC we have seen for an SZ gene identifier. I’m also running the program on the autism positives right now, and it appears that the AUC is running at about similar levels to where they were with E1 and E2.

Krishnan, A. et al.Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nature Neuroscience19,1454 (2016).
I think we are at the point where we will begin writing the paper. These results are a satisfying in that it’s probably just the nature of SZ that genes implicated are going to be difficult and somewhat disparately related. That’s not necessarily a bad thing. Polygenic positives are by definition only contributing slightly towards a disease. The strongest p-value for a SNP in an SZ GWAS has a frequency of 0.765 in SZ participants and a frequency of 0.75 in control participants. A low AUC in an SZ gene labeler is almost expected.
Goals for next week: Write the paper
Week 3: Processing Data from FireBrowse and PathLinker
On Monday we all met with Anna to agree on some goals for the week. Kathy and I set out to acheive several things: A. to figure out how to download data from FireBrowse on colon adenocarcinomas, to perform several statistical analyses on that data and to visualize it in some form using pylab, B. to write a code that produced informative graphs of the different signaling pathways from text files containing information about the edges and nodes and C. to implement PathLinker and LocPL and to create graphs of the top k paths on GraphSpace.
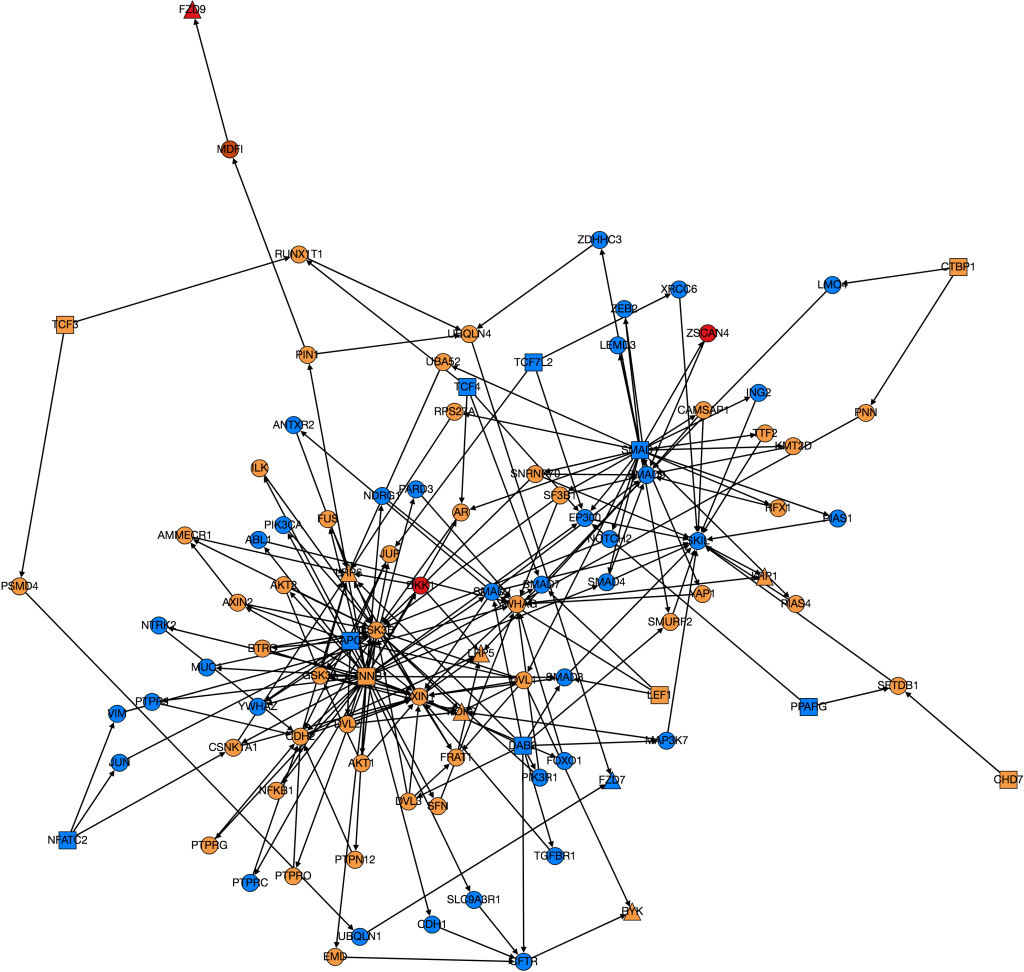
Originally to acheive our first goal, we tried to use Nicholas Egan’s pepper pathway code to download and format the data. However, while we were able to decipher how it worked, it ended up not being all that useful to us, so Kathy downloaded all the gene files related to colon adenocarcinoma onto her computer . Unfortunately, we’re still not really sure how to use them. We wrote a code that took text files containing information on edges and nodes and spit out a graph with color-coded nodes that were labeled with the node name and type and edges that were labeled with the type of interaction that was occuring between the two nodes for example “physical” or “phosphorylation”. I have attached to examples of the graphs we produced below, one very complex and the other very simple.
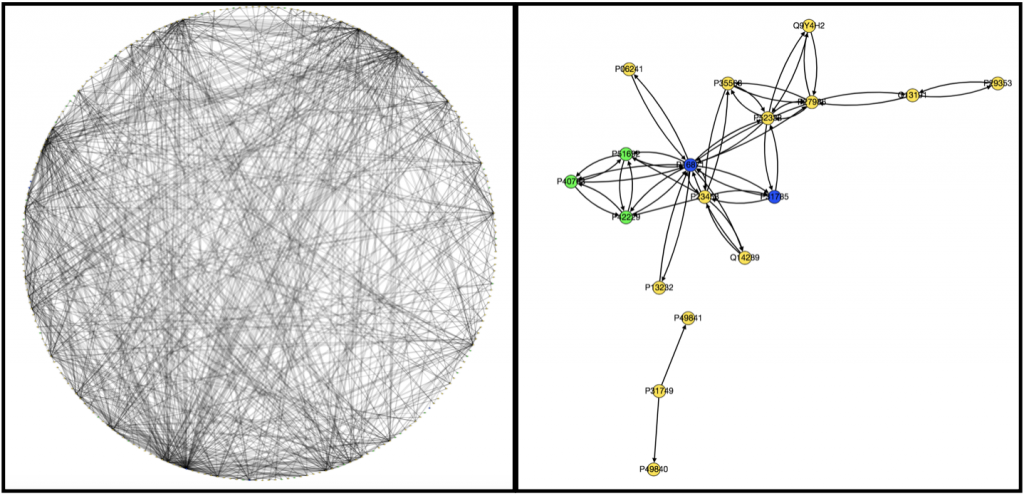
We also ran PathLinker on the Wnt pathway and made a graph of the top 200 best paths produced.
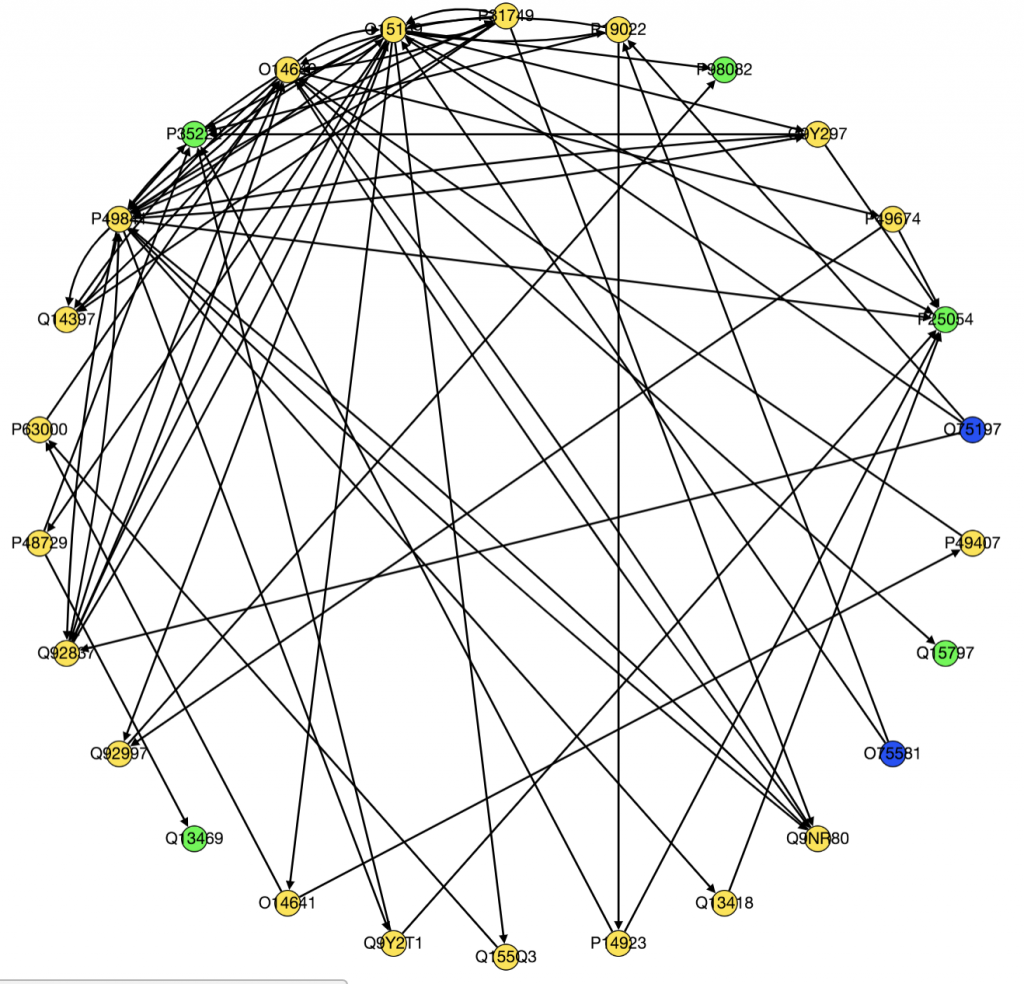
Moving forward we have several goals. Kathy is trying to figure out how to implement LocPL. I am still trying to figure out how to process and use the FireBrowse data. Because I will need to integrate methylation data from FireBrowse in my project, it is essential I figure out how to use it.
Overall, this week I have learned a lot about data processing, writing code to read text files, and using NetPath, PathLinker, FireBrowse, and GraphSpace. These skills should come in handy as I move forward in my project.