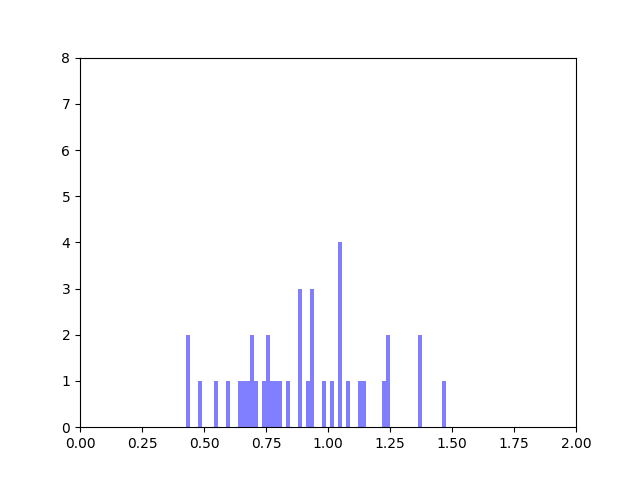
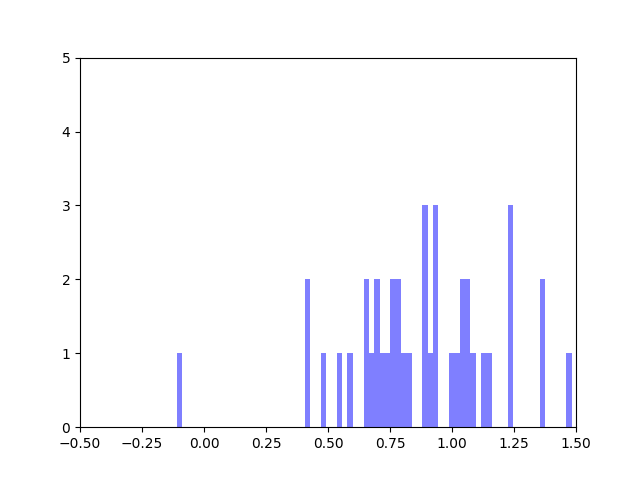
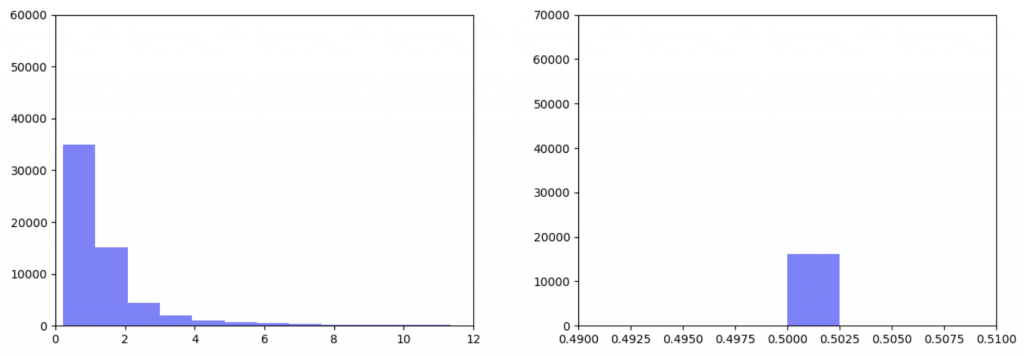
In my last blog post, I talked about how I was concerned about how small the range of normalized node scores is. This week I’ve been trying to figure out why that is. To do this I’ve been making histograms of each step of the process from foldchange to Xv to Cv. This is an example of that process for one gene:
Distribution of foldchanges across patient samples for a single gene.Distribution of Xvs across patient samples for a single gene
The Cv of the gene above was 0.5000000003. Unfortunately, it looks like a lot of genes even with fairly different fold change and Xv distributions end up with very similar Cvs.
Ibrahim realized that this is occurring because there is an error in the equations we were using so we will have to rethink the way we normalize the data.
So now we are in the experimental validation phase. This week, we ran a trial run of the cell tracking software, using newly cultured cells from a drosophila melanogaster, or fruit fly, line. These lines are suitable for validating that the genes are involved in cell motility due to the high degree of conservation between humans and fruit flies in basic cellular mechanisms.
This week, I’ve been creating a tabular file with all TCGA-COAD samples at the top of the file as column names, with genes at the sides. I should have a 512 x 60484 matrix file when done. However, with Sol’s help I realized that the file I initially output was actually 512 samples at the top, with only 443 columns after, but still with 60484 lines to the file.
Therefore, I think there’s something wrong in how I’m categorizing/organizing the samples.
This week Kathy, Usman and I met to discuss how we would combine the projects we’ve been working on into a cohesive pathway and how we would analyze the output of CancerLinker as compared to PathLinker.
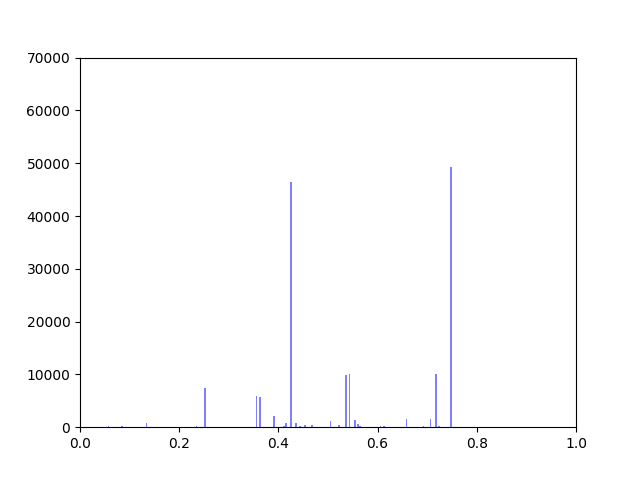
I have been working on ways to visualize the data. One thing I wanted to look at was how incorporating gene expression data would change the overall distribution of edge weights in the interactome.
The original interactome has a reasonable distribution with two values that seem to appear frequently around 0.4 and 0.8.
Original Interactome
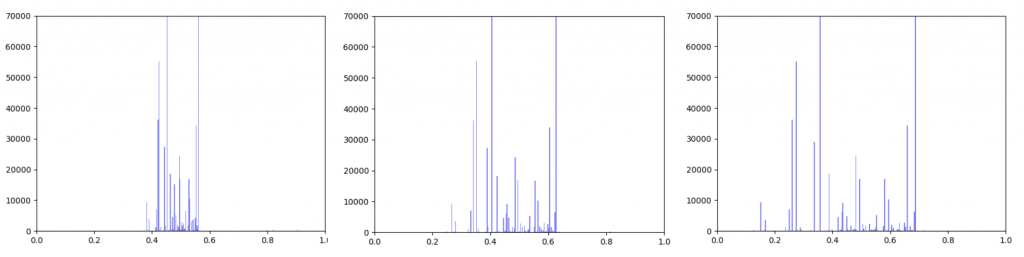
Gene expression data was incorporated into the original interactome with a beta value. The beta value determines the weight of the original edge weight when including the gene expression data. So the higher the beta score, the lower the importance of the gene expression data. I made three histograms one for a beta=0.25, one for beta = 0.5 (equal contribution between original edge weight and gene expression data) and one for 0.75
Distribution of edge weights with gene expression incorporated. Left B=0.25, Middle B=0.5, Right B=0.75
As can be seen in the histograms above, when gene expression data is weighted more heavily, edges weights are more closely clustered. To investigate why this occurred I made two additional histograms: one of the gene expression data before it was transformed and one of the gene expression data after it was transformed.
Left: Gene expression data before transformation Right: Gene expression data after being transformed
After the gene expression data is transformed, there is extremely little variation. Additionally all the gene expression data is greater than or equal to 0.5 which should be the median. This would explain why weighting the gene expression data more heavily causes more closely-clustered edge weights. I’m not sure how to fix this. It seems like an error, but I’ve been over the code multiple times and the math seems right to me. So my next step is to figure out what’s going on there.
In the meantime I took the output from the pipeline that Kathy put together and put graphs for the top 1000 Wnt paths with β=0.25, 0.5 and 0.75 up on GraphSpace. If I figure out what’s wrong with the function that transforms the edgeweights, I will run it again and re-upload the updated graphs.
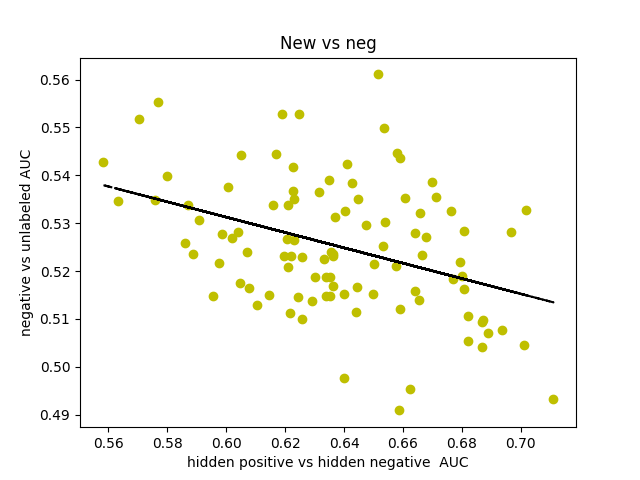
I was curious about what our AUC would look like if instead of comparing hidden positives to unlabeled, we compared hidden positives to hidden negatives, wondering if that would get us a more valid representation of the network’s ability to predict genes associated with schizophrenia. What if the unlabeled genes ranked above the hidden positives were simply actual centers of genetic perturbations in schizophrenia?
Oddly enough, the scores actually decreased from 0.65-0.70 to 0.60-0.65. I found that the AUCs for comparing hidden negatives to unlabeled nodes hovered around 0.53, meaning that the hidden negatives were ranked mostly in the top half of the list.
This somewhat makes sense though. The basis for the negative set is that they are genes in non-neurological diseases, but that doesn’t necessarily exclude them from being included in a more subtle, polygenic disorder that’s dependent on additive effects of hundreds of genetic perturbations. It also makes sense that genes involved in other diseases might have a slightly higher probability of being associated with some other disease.
I decided to make a new negative set. I started by gathering every single node in the 0.150 network. There is a resource called SZGR 2.0 (https://bioinfo.uth.edu/SZGR/) that collects all sorts of evidence for genes being associated with schizophrenia. Using this list, I excluded any gene that had any evidence for being associated with schizophrenia. I then took differential gene expression data from autism spectrum disorder, bipolar disorder, and major depressive disorder as well as schizophrenia (http://science.sciencemag.org/content/359/6376/693). If a gene was not differentially expressed in any of these diseases (FDR>0.5 for schizophrenia, FDR>0.2 for others) and was in the other list, I added that gene to my list of negatives, which was 1561 genes long. I found that this list, when hidden negatives were compared to unlabeled genes, had an AUC of about 0.44, which means that our negatives are finally oriented around schizophrenia rather than non-neurological diseases. I also found that the AUC for hidden positives vs hidden negatives spiked up to 0.75, meaning that our program is legitimately good at predicting genes associated with schizophrenia.
I found no significant performance differences from factoring in evidence levels.
This week I began to write code to weight the Pathlinker Interactome using the Bayesian Weighting Scheme. This will be useful to compare the HIPPIE and the Pathlinker Interactome but it also serves as a check to confirm whether the code accurately weights the interactomes. The Pathlinker Interactome was initially weighted using the same scheme and so the weights generated should match the pre-existing weights of the interactome. Next steps will involve comparing the two interactomes.
I spent some time writing code that allows conversion of a list of Uniprot IDs to the common name of the genes. This code will probably prove useful at some later point. I extended this to convert from Ensemble IDS to Uniprot IDs as well. However, the Ensemble and Uniprot databases do not entirely overlap and so there are some Ensemble IDs with no corresponding Uniprot IDs. I currently have no ideas about how to solve this problem but it does not appear to be one that needs to be urgently solved.
Changed our SZ positives to be evidence based. The number of layers the positives are in is determined by the evidence level. It does better than before. We are confident in the gene lists and are looking into the candidate genes for experimental validation
Last week, we were lucky enough to attend the GCC/BOSC conference hosted at Reed. Although I spent the majority of the week volunteering and attending sessions, we also had a deadline for a two-page extended abstract of our CNB-MAC paper to be submitted to the main conference proceedings. We successfully cut down our original paper into two pages and submitted the abstract on Friday.
As of now, we’re almost ready to dig into the experimental portion of this project, but there are still a few things we need to iron out. At the end of last week, Alex noticed a “bug” in how we’ve been calculating the AUC of our multi-layer algorithm. Therefore, one of our priorities this week is to find a more accurate AUC calculation that better represents our method. Once that is completed, we will move on to assembling a list of candidates for experimentally validation. Hopefully by the end of the week we will have chosen 8-10 candidates that are not only biologically interesting but conserved in Drosophila as well.
This week I spent the majority of my time at the GCC/BOSC Conference both volunteering at and attending talks. Most of the talks concerned the software suite Galaxy and how people were adapting it or extending it for different use cases and new applications. Not having used Galaxy before nor intending to, these talks did not prove very useful to me. There were some talks that concerned more general biological problems and issues within the scientific community. These were accessible to a wider audience and I was able to learn some aspects of statistics and experimental design from them. This included talks on the importance of talking to the community of people who have the particular condition being studied as well as some concerning best practices on handling data.
I spent some portion of time fixing minor bugs in my code for the Bayesian Weighting Scheme. As a follow-up to a previous post, the run time of the code is now down to 2 or 3 minutes at the most. Using sets and tuples reduced the run time by several orders of magnitude and some other time saving measures also contributed to this much lower number. This is now sufficiently within the time frame of the CREU so the code will probably need much more speeding up. Next steps involve changing the code to weight the PathLinker Interactome to check if the code works accurately as well as parsing through the math carefully.
This week we spent most of our time attending and volunteering at GCC/BOSC (Galaxy Community Conference/ Bioinformatics Open Source Conference). While the conference wasn’t extremely relevant to what we’re working on right now, we learned how to use software like Galaxy and Intermine, which might be useful to us in future projects. Also, this was the first conference I’ve ever attended so it was very interesting to learn how conferences work and to meet people.