My name is Amy Rose, and I’m a post-bac in Anna’s lab this summer. I graduated last month with an Alt. Biology degree with an emphasis in Computer Science. Taking Anna’s classes in my first two years at Reed was the start of my interest in computational bio. I spent my junior year studying computer science at The University of Sussex, and after this summer I will be starting as a software engineer at Puppet here in Portland.
When it came time to find a thesis project, I thought it would be interesting to explore an area of biology that I hadn’t had time to study while at Reed. I was coadvised by Anna and Sam Fey, who is an ecologist. Sam’s research on thermal variation led me to my project, which focused on modeling the effect of thermal variation on freshwater phytoplankton using real world data.
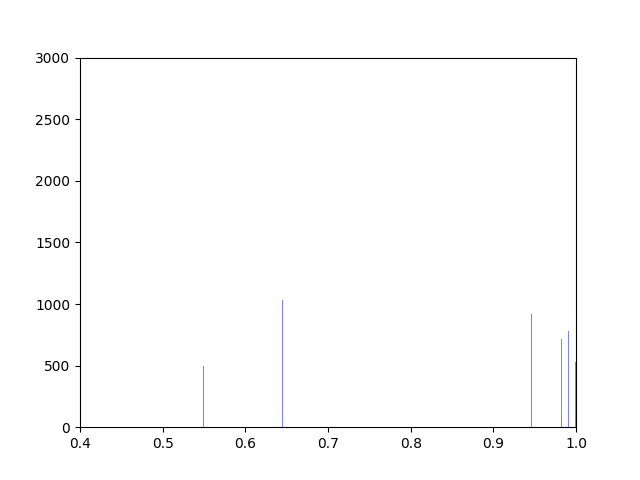
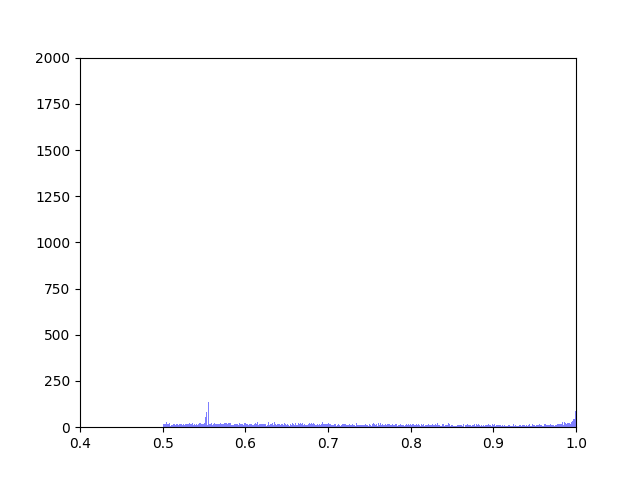
Phytoplankton are ectothermic, which means that they are not able to regulate their own body temperature. Additionally, due to their small size it is difficult to empirically measure the variance in their body temperature due to movement through thermally variable environments. My thesis began to resolve the impact on movement on body temperature and fitness. In this context, fitness represents the overall change in population size of phytoplankton based on temperature-dependent birth and mortality rates.
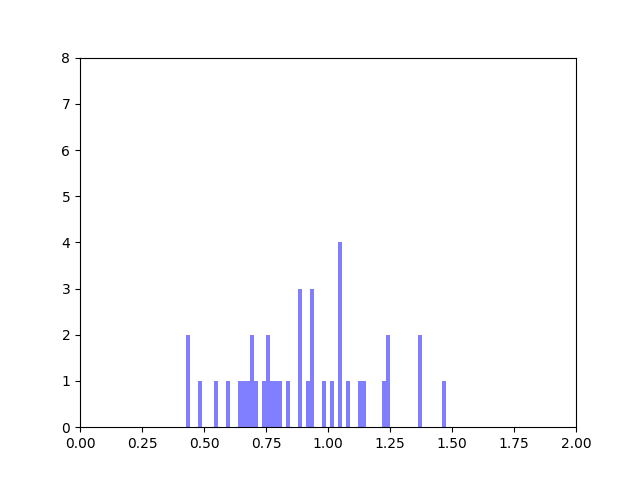
Temperature data was collected from Sparkling Lake in Vilas County, Wisconsin at intervals from .5 to 3m throughout the lake with a frequency as high as every minute over a period of 26 years. We interpolated the collected data to fill in estimated temperatures over depths which were not collected, as seen in the figure below.

We created five algorithms representing different theoretical patterns of phytoplankton movement throughout the water column, which we plotted against the data. This gave us a framework to understand the limits of what body temperatures phytoplankton may be experiencing. The second stage of the project was to plot these simulated body temperatures against a function representing phytoplankton fitness.
This summer, we hope to extend my thesis research over space and time. For my thesis, we focused on a single season, but we’re currently looking at extending the movement algorithms over all 26 years of data. We’re also interested in exploring more datasets sourced from lakes in different geographical locations. Additionally, we’re analyzing the effects of changes to the fitness function.